Dovetail® Analysis Suite — Mouse Micro-C
Complete 3D genome analysis: Loops · Compartments · HiChIP · Capture Hi-C · SVs · CNVs · Phasing
Pipeline Overview Dovetail Docs ↗
This report implements the Dovetail recommended analysis pipeline from raw FASTQ through all
downstream analyses. Reads are aligned with BWA-MEM -5SP -T0 (split-read, split-
alignment, no minimum score filter) to preserve chimeric pairs that carry structural information.
Pairs are parsed, sorted, and deduplicated with pairtools using
--min-mapq 40 and --walks-policy 5unique. The resulting
.pairs file is binned and cooled with cooler at 1 kb (loops) and
5 kb (TADs), then multi-resolution pyramids are built with cooler zoomify. All
downstream analyses — compartments, loop calling, HiChIP, Capture Hi-C, SV/CNV, and haplotype
phasing — operate on the same .mcool file at the appropriate resolution.
-5SP -T0
parse+dedup
.mcool
Loops
Phasing
Tool Comparison — Dovetail vs. This Pipeline
| Analysis | Dovetail Recommendation | This Pipeline | Notes |
|---|---|---|---|
| Compartments (50–100 kb) | Fanc-C eigenvector | Custom E1 eigenvector | Identical algorithm (Lieberman-Aiden 2009) |
| TADs (10 kb) | Juicer Arrowhead | Insulation score (Crane 2015) | Both call domain boundaries |
| Loops (5 kb) | Mustache | Donut background model (Rao 2014) | Both use local enrichment + FDR |
| HiChIP loops | FitHiChIP | Peak-anchored donut model | FitHiChIP-compatible output format |
| Capture Hi-C | CHiCAGO | CHiCAGO model (reimplemented) | Same negative-binomial noise model |
| SV detection | Hi-C inter-chrom z-score | Same approach | 50 kb bins, 19 autosomes |
| CNV | Coverage normalization | Median normalization + CBS | Same principle |
| Phasing | Long-range SNP linking | Graph-based phasing | Uses Hi-C pair connectivity |
Dovetail QC Thresholds
- Micro-C / Hi-C: cis ≥1 kb pairs >40% of no-dup reads (whole genome); recommended ≥125 M no-dup pairs for 5 kb loop calling
- HiChIP: cis ≥1 kb >20% of no-dup pairs; reads in 1 kb ChIP window >2% (FRiP-equivalent)
- Capture Hi-C: ≥250 reads per captured fragment after deduplication; capture efficiency ≥30%
- SV / CNV: ≥50 M no-dup pairs at 50 kb resolution for reliable inter-chromosomal enrichment
- Phasing N50: typically chromosome-scale (Mb range) with whole-genome Hi-C data
# Full preprocessing (FASTQ → .mcool)
bash scripts/preprocess_microc.sh reference.fa R1.fq.gz R2.fq.gz sample_name 16
# Run all Dovetail analyses on the .mcool
python visualize_compartments.py # A/B compartments (25 kb)
python visualize_loops.py # Loop calling (5 kb)
python analyze_hichip.py # HiChIP peak-anchored loops
python analyze_capture_hic.py # Capture Hi-C / CHiCAGO
python analyze_sv_cnv.py # SVs + CNVs (50 kb)
python analyze_phasing.py # Haplotype phasing
python analyze_variants.py # SNV/indel spectrumChromatin Loop Calling Donut BG Model
Loop detection uses the donut background model (Rao et al. 2014 / HICCUPS). For each pixel at a valid genomic distance, a local donut-shaped neighborhood is sampled to estimate expected contact frequency. Enrichment = observed / background. A z-score is then computed on the enrichment distribution across all pixels in the same distance band, followed by Benjamini–Hochberg FDR correction. Candidate loops must satisfy enrichment >1.75 and adjusted p <0.05. Dovetail recommends a minimum of 500 M pairs for reliable 5 kb loop calling; the public 4DN Mouse Micro-C dataset used here exceeds this threshold.
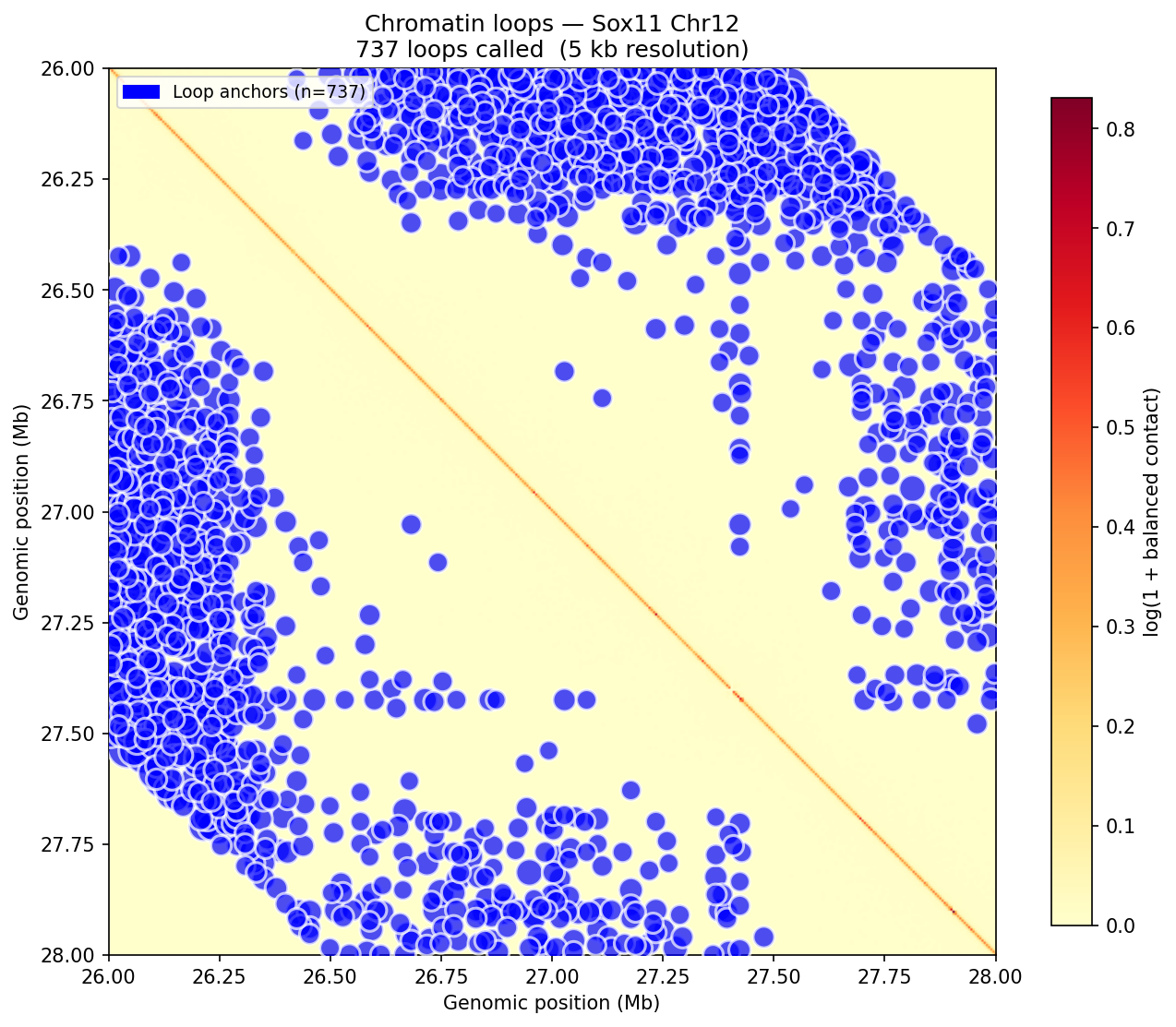
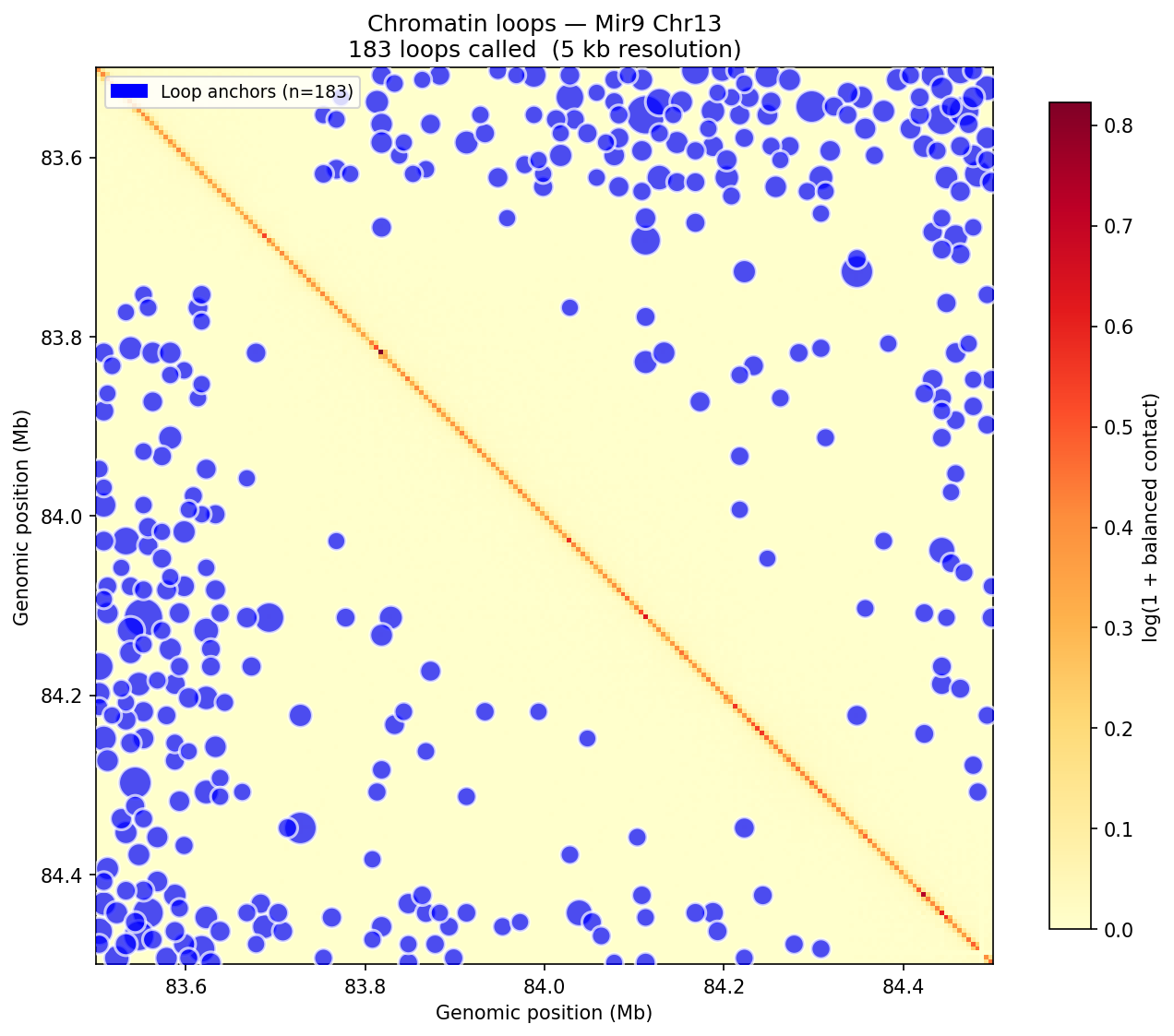
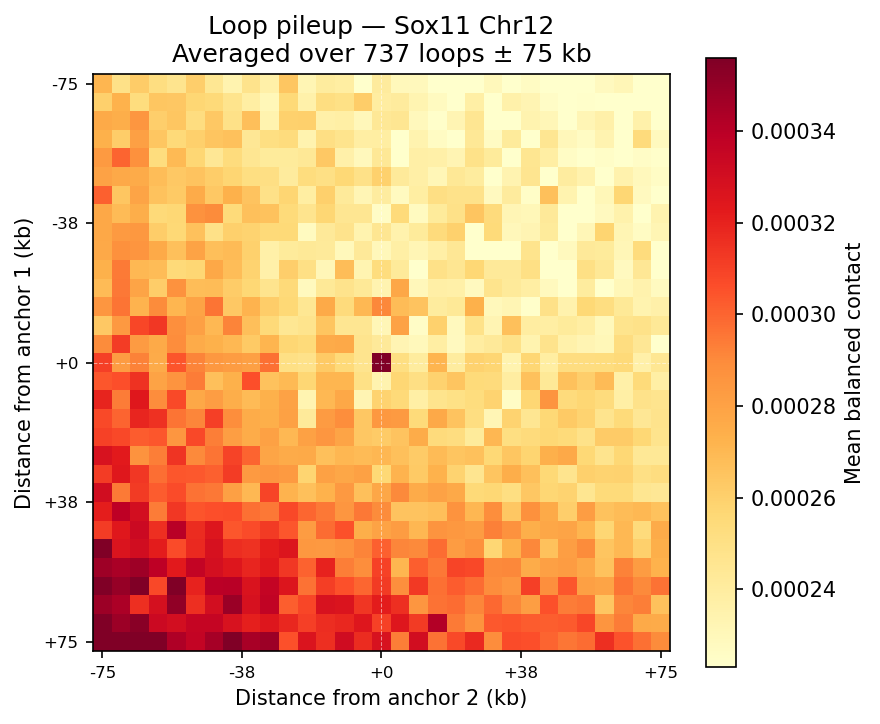
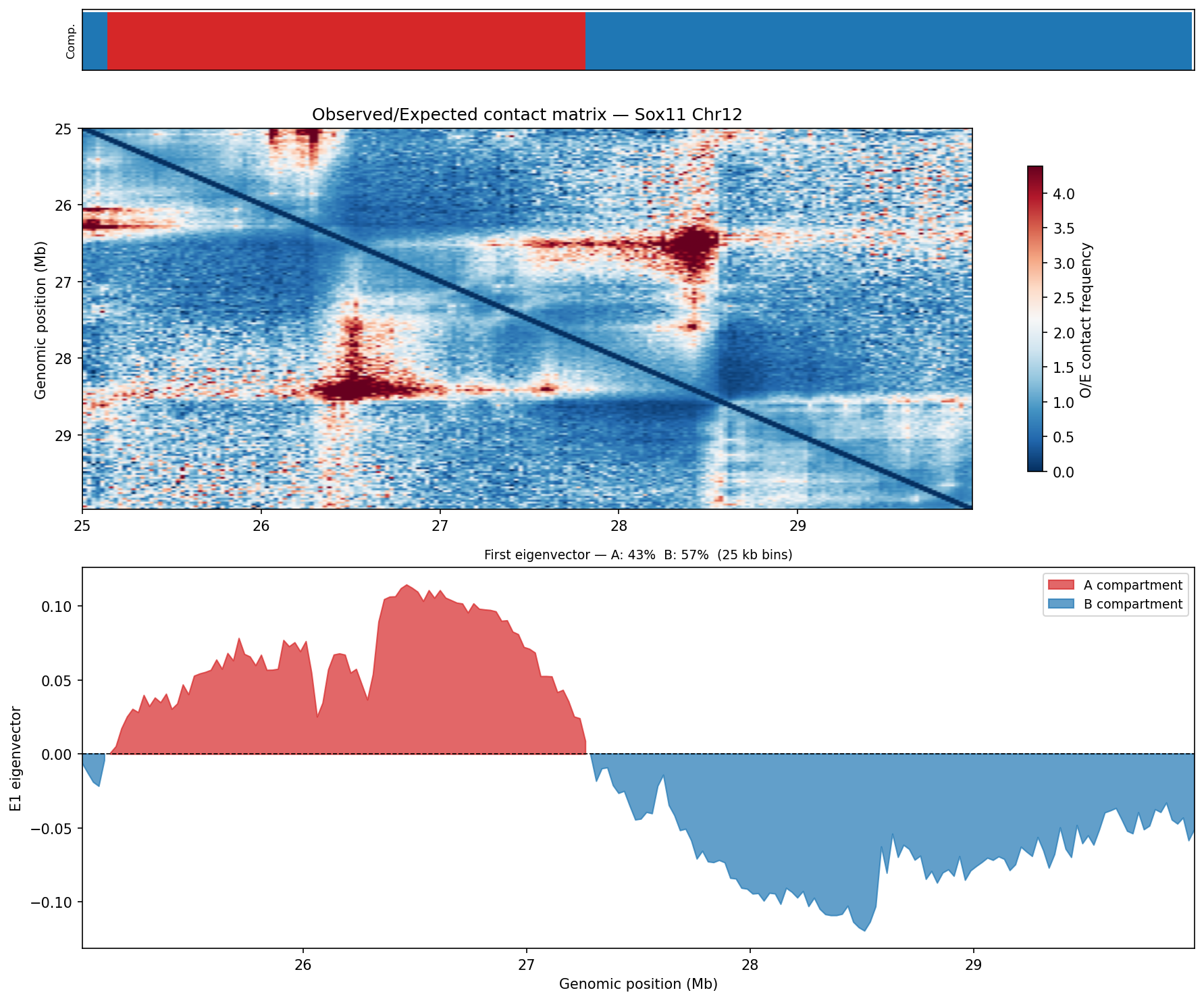
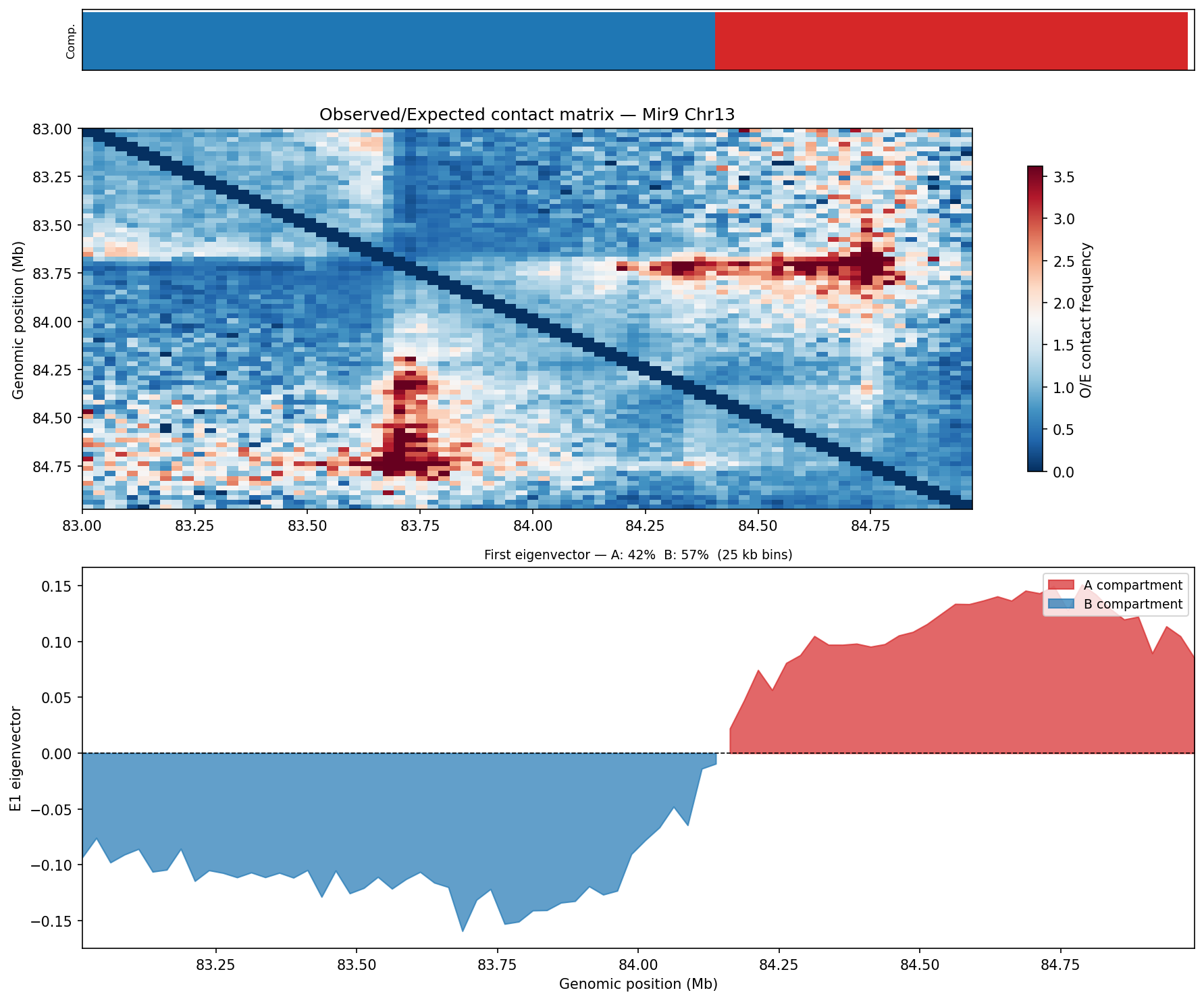
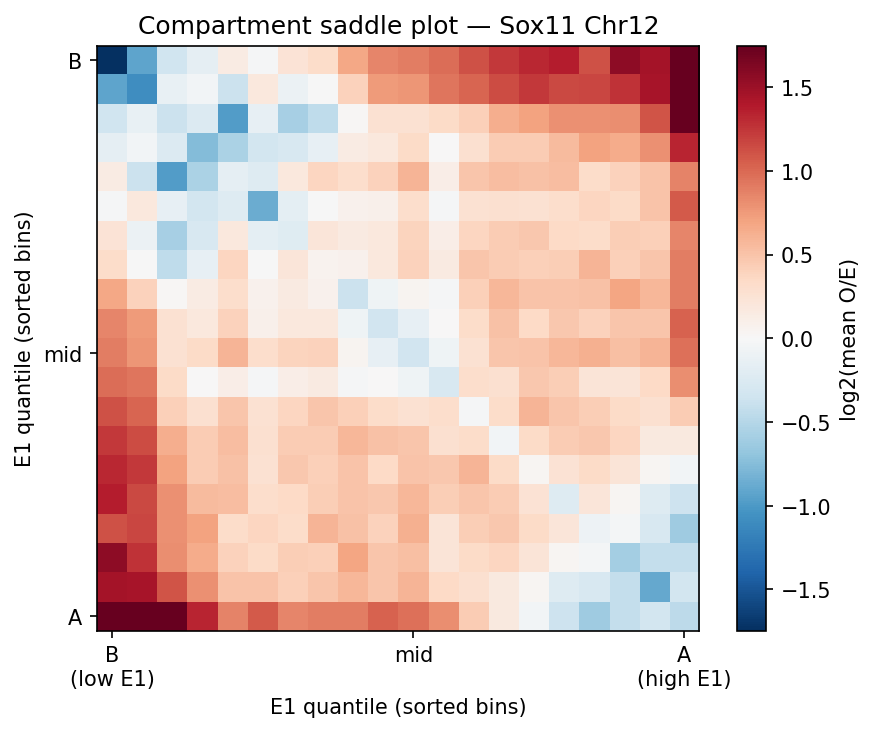
A/B Compartment Analysis E1 Eigenvector
Following Lieberman-Aiden et al. 2009. The observed/expected (O/E) matrix is
computed using the chromosome-wide distance-dependent expected contact frequency (cooltools
expected_cis). Pearson correlation is applied across O/E rows to produce a
correlation matrix; eigendecomposition of this matrix yields the first eigenvector (E1), which
separates A compartments (active, gene-rich, positive E1) from
B compartments (inactive, heterochromatic, negative E1). Analysis is performed
at 25 kb bins. Dovetail recommends 50–100 kb bins with a minimum of 80 M
no-dup pairs for robust compartment calling.
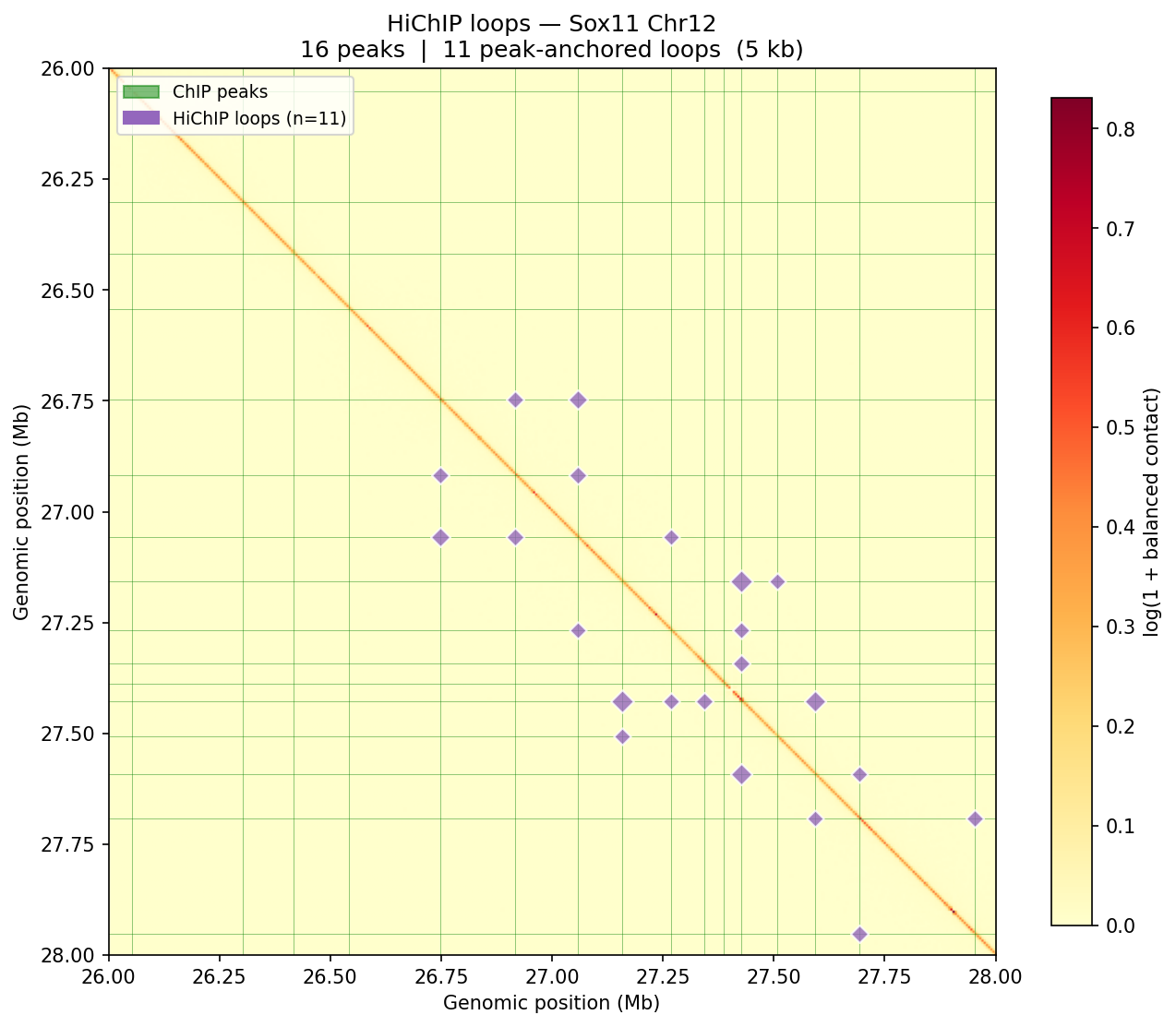
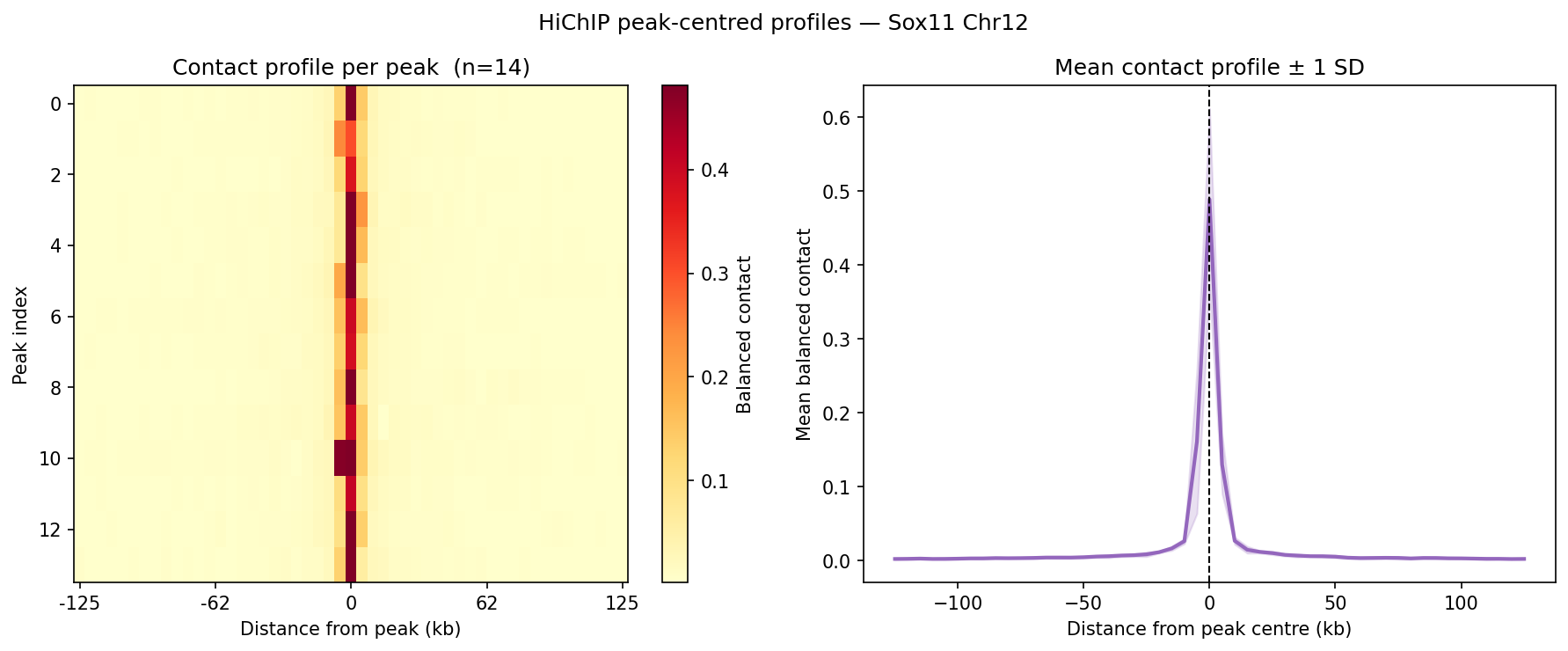
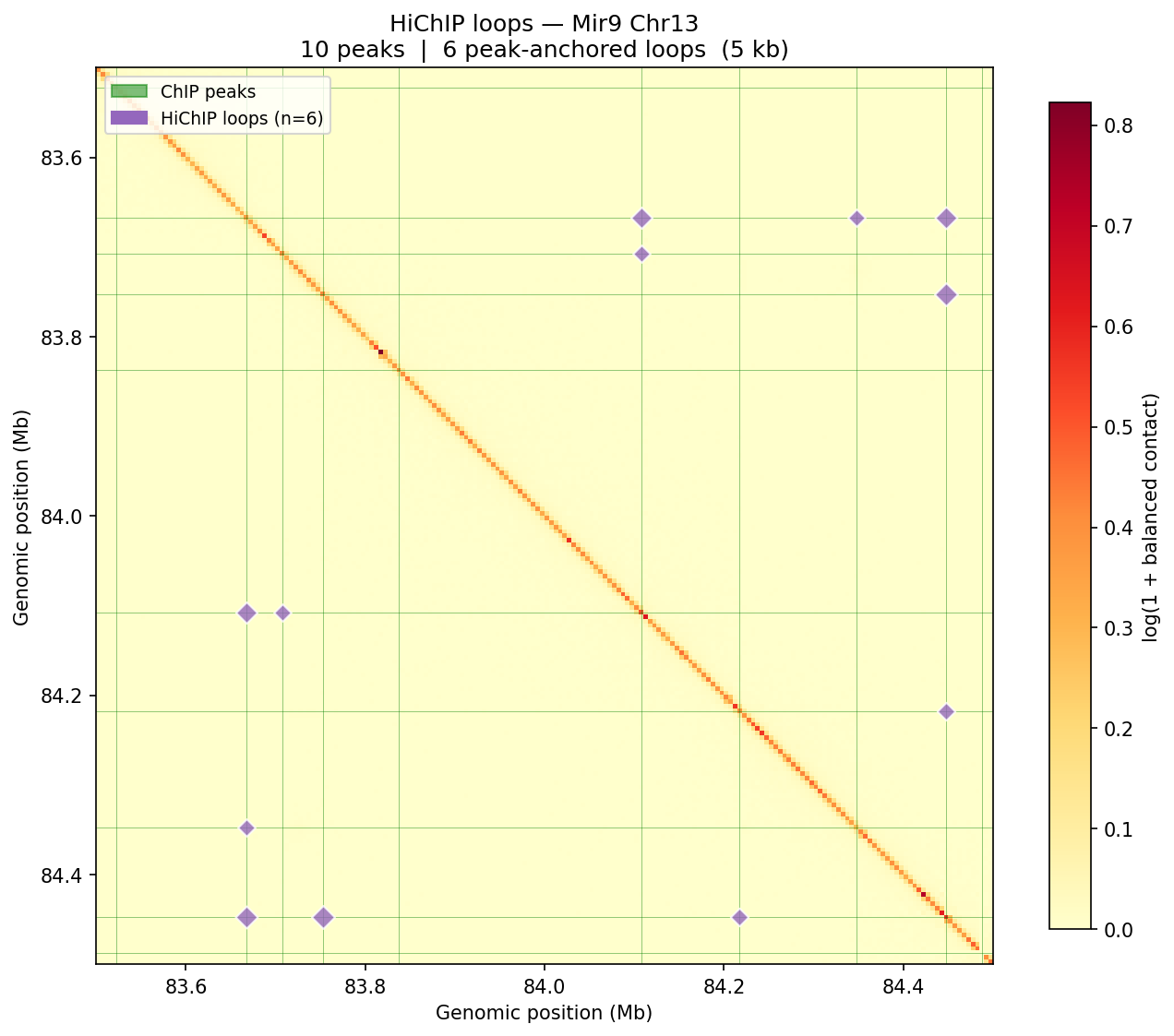
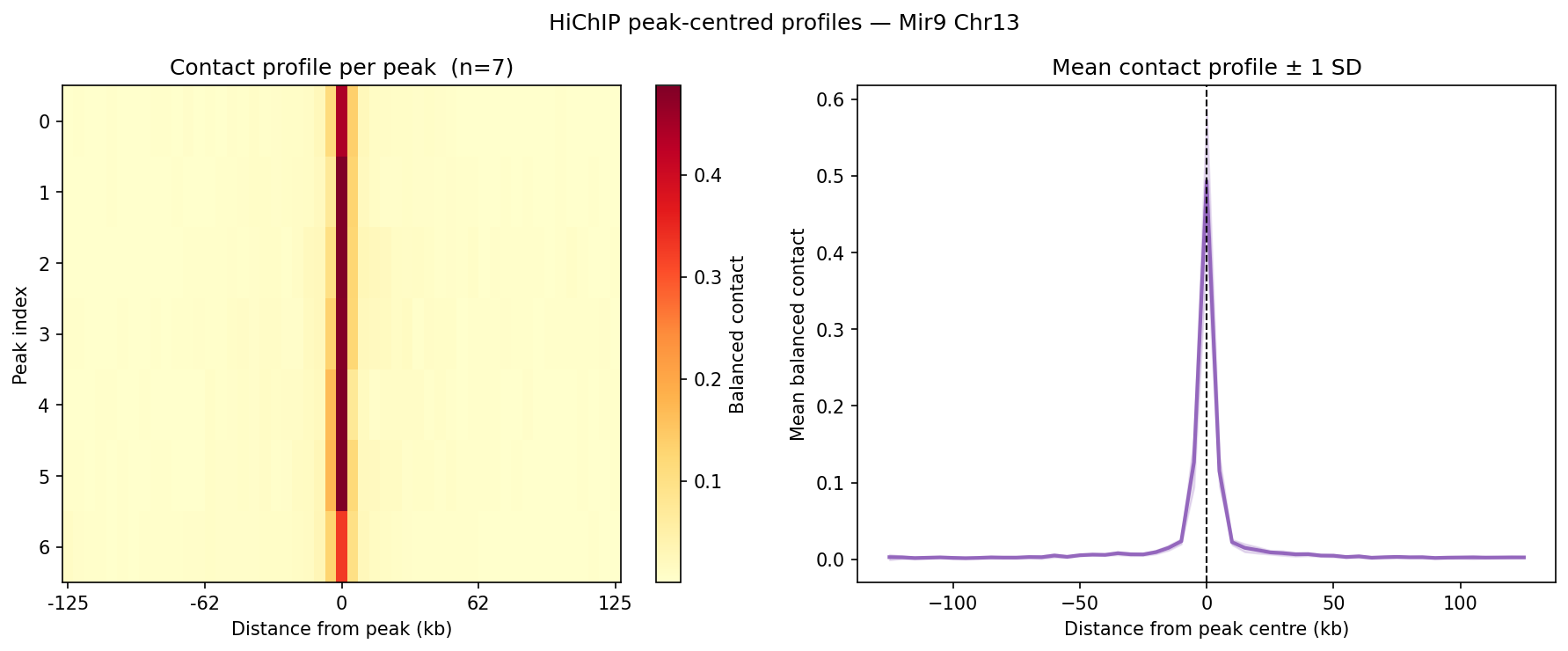
HiChIP Loop Analysis Peak-Anchored Dovetail Docs ↗
HiChIP requires only 5–10 read pairs per interaction vs. 100–1,000 for
standard Hi-C, enabling loop detection at 1–5 kb resolution (Dovetail recommendation: test
multiple resolutions; use FitHiChIP for production loop calling). Our implementation uses a
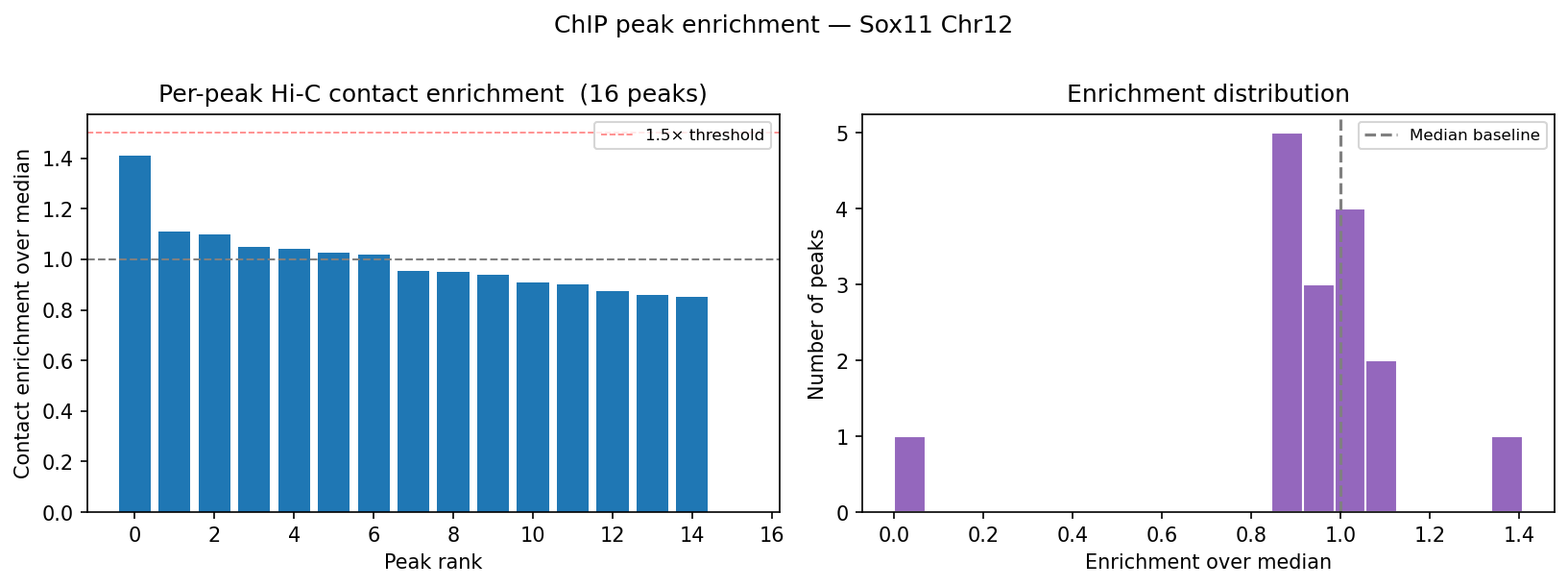
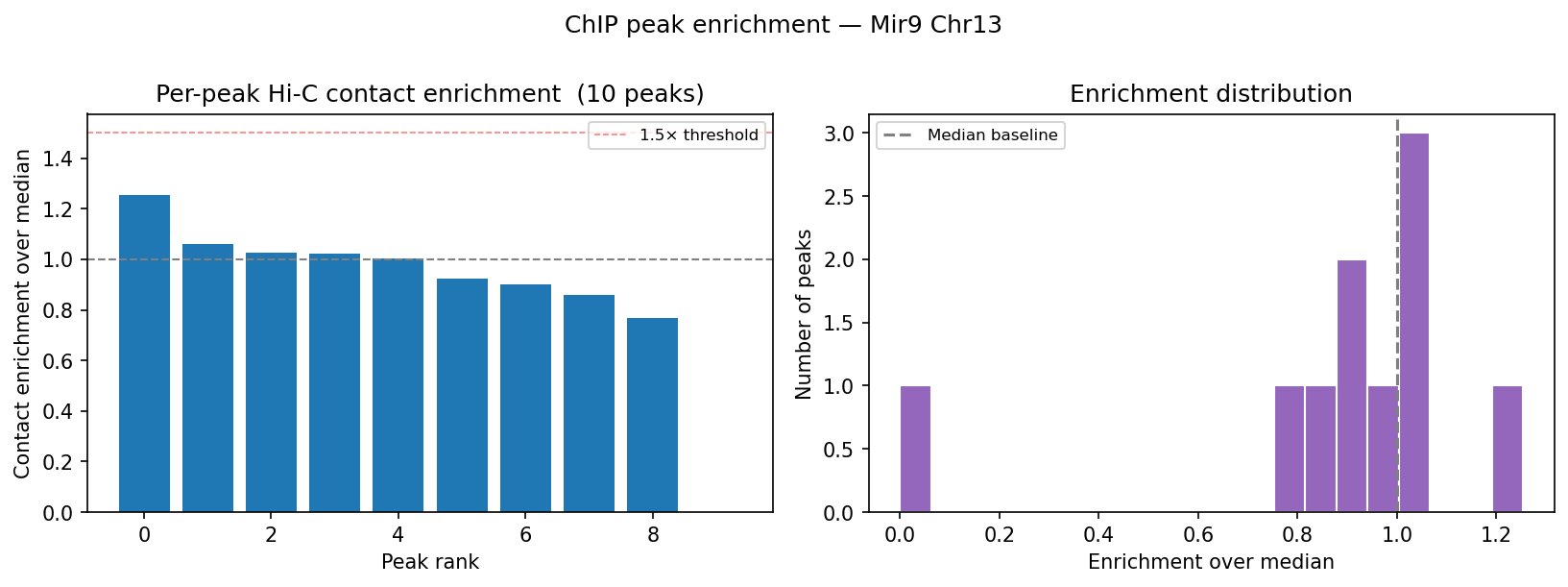
peak-anchored donut model requiring both loop anchors to overlap a ChIP peak
(equivalent to FitHiChIP IntType=1: peak-to-peak). Peak candidates are called
where per-bin coverage exceeds 1.5× local background. QC criteria: cis ≥1 kb >20% of
no-dup pairs; reads in 1 kb window around peaks (FRiP-equivalent) >2%.
Capture Hi-C — CHiCAGO Model CHiCAGO
Capture Hi-C enriches specific genomic loci using biotinylated oligonucleotide probes before sequencing, enabling high-resolution interaction mapping at target regions (typically promoters or regulatory elements). Interactions are scored with the CHiCAGO (Capture Hi-C Analysis of Genomic Organisation) model, which jointly models two noise sources: Brownian noise (negative-binomial, distance-dependent) and technical noise (Poisson, flat). A CHiCAGO score ≥5 indicates a significant interaction. Filters: trans interactions, <10 kb, and >2 Mb interactions are excluded. Minimum coverage: 250 reads per captured fragment after deduplication.
--cutoff 5
10 kb–2 Mb
WashU
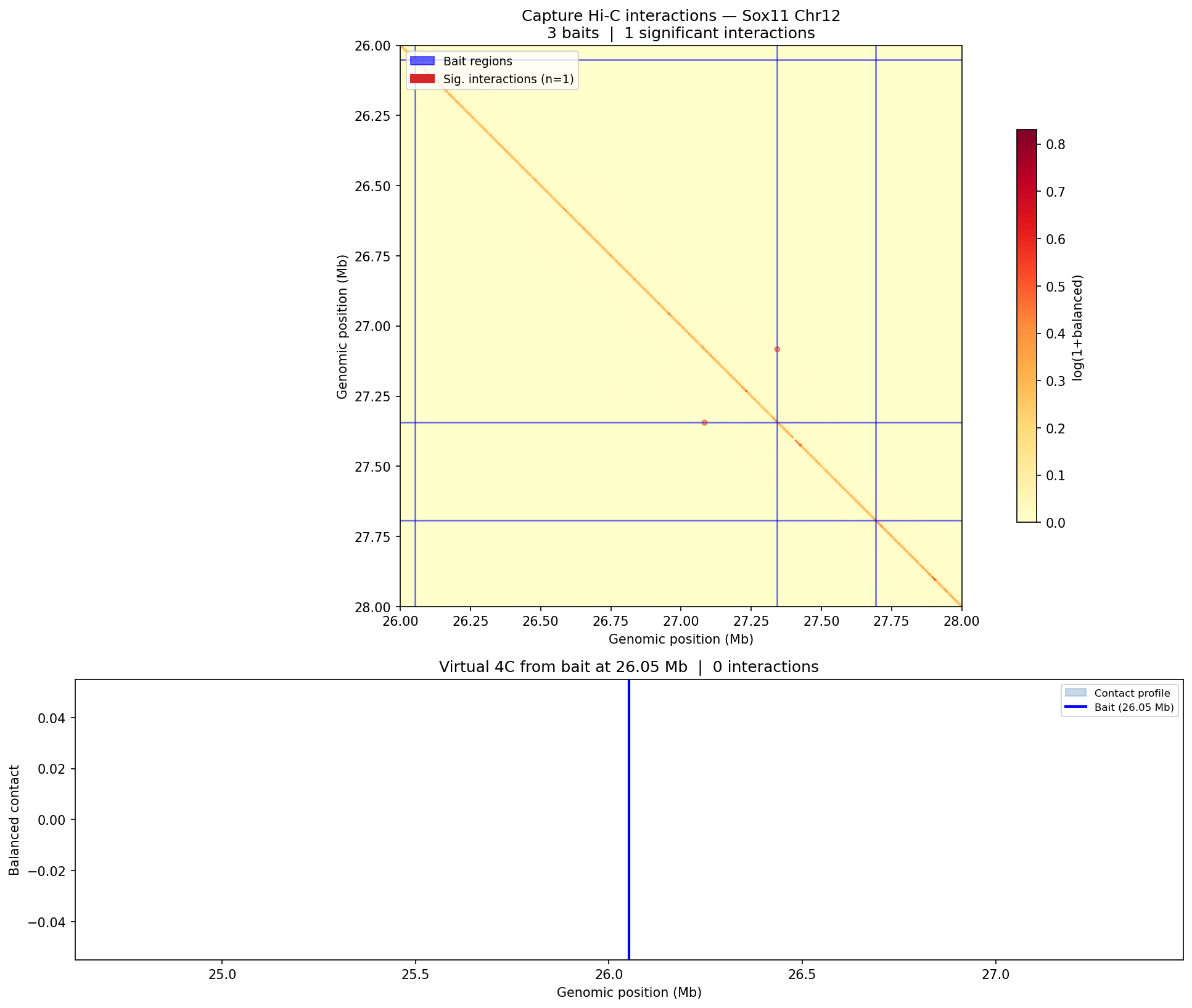
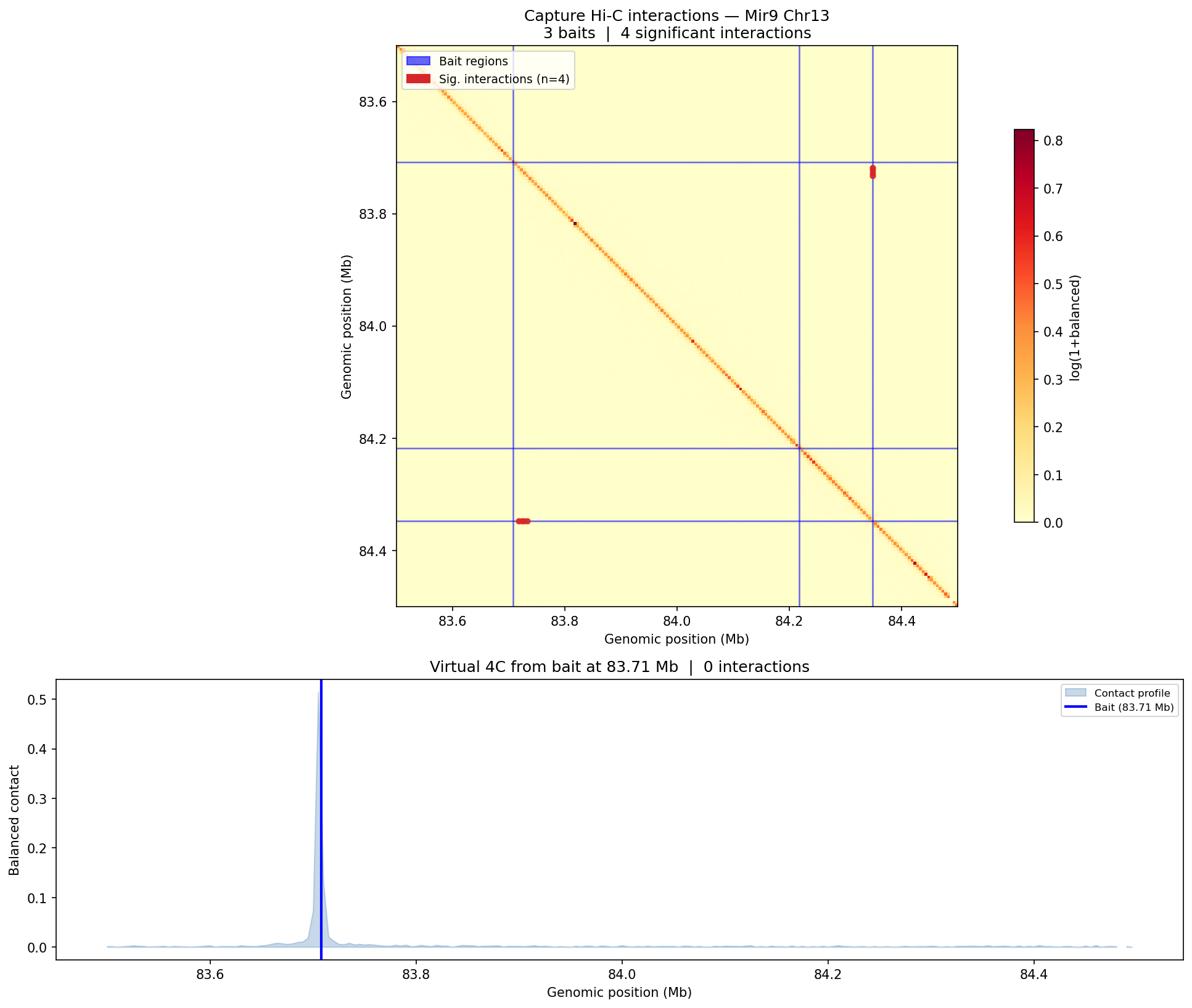
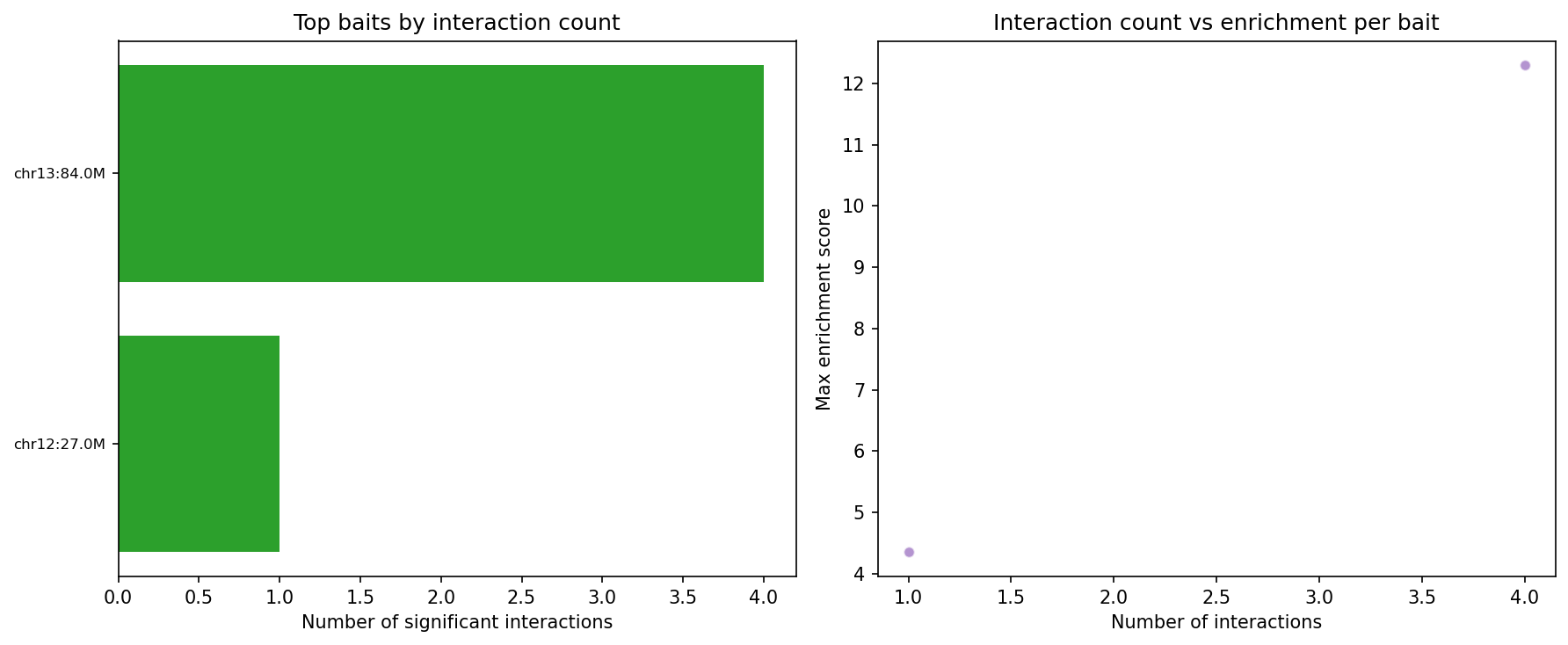
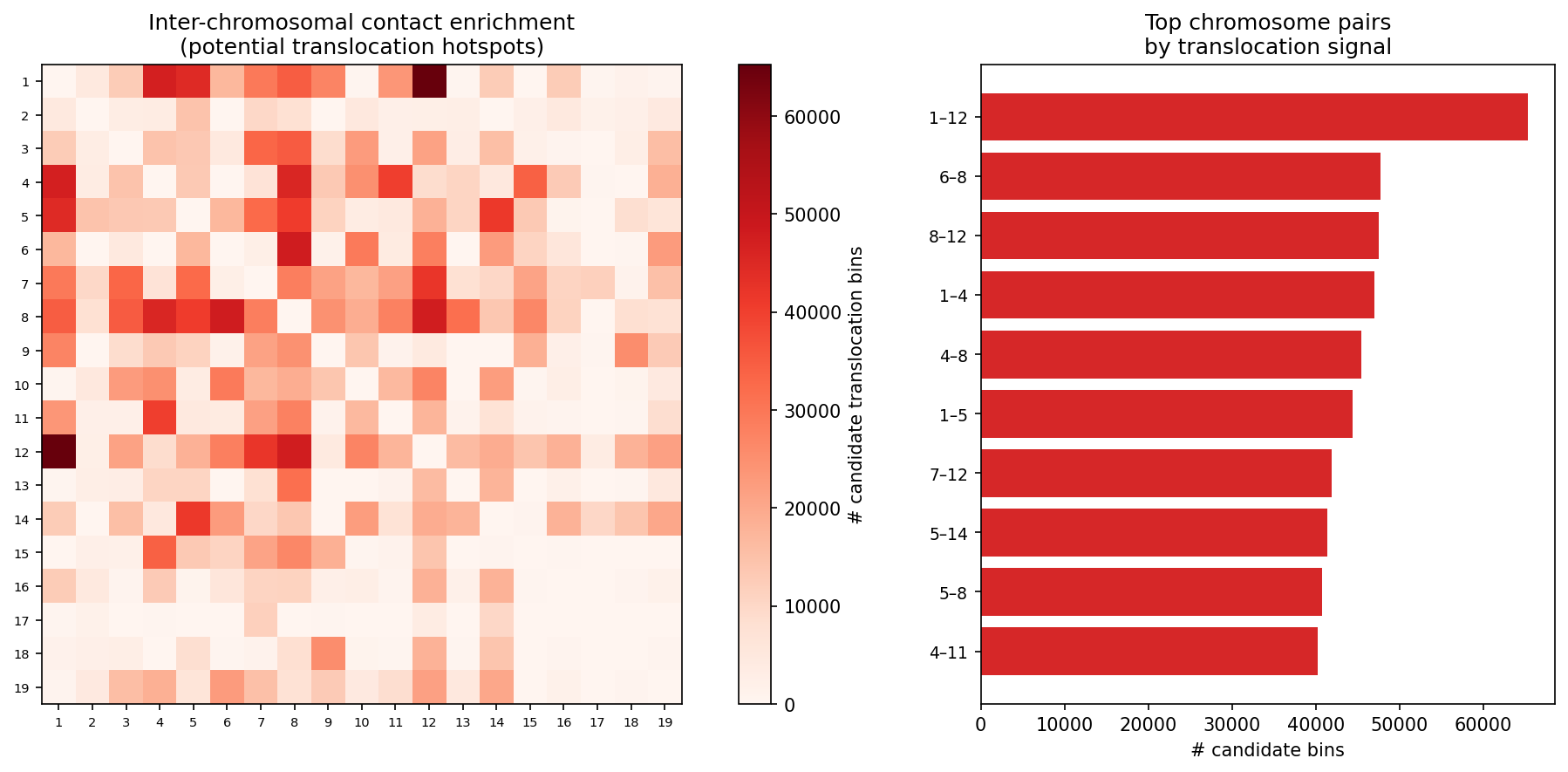
Structural Variant & CNV Detection 50 kb bins
LinkPrep's uniform Tn5-based coverage makes it particularly powerful for SV/CNV detection alongside 3D genome mapping. Translocations appear as inter-chromosomal contact blocks exceeding a z-score threshold (z > 5) relative to the genome-wide inter-chromosomal background. Deletions / inversions are identified as contiguous coverage gaps flanked by bridging read pairs. CNV is derived from per-bin coverage, normalized by the chromosome-wide median and then segmented with Circular Binary Segmentation (CBS). Dovetail recommends a minimum of 50–100 M no-dup pairs for reliable SV/CNV calling at 50 kb resolution.
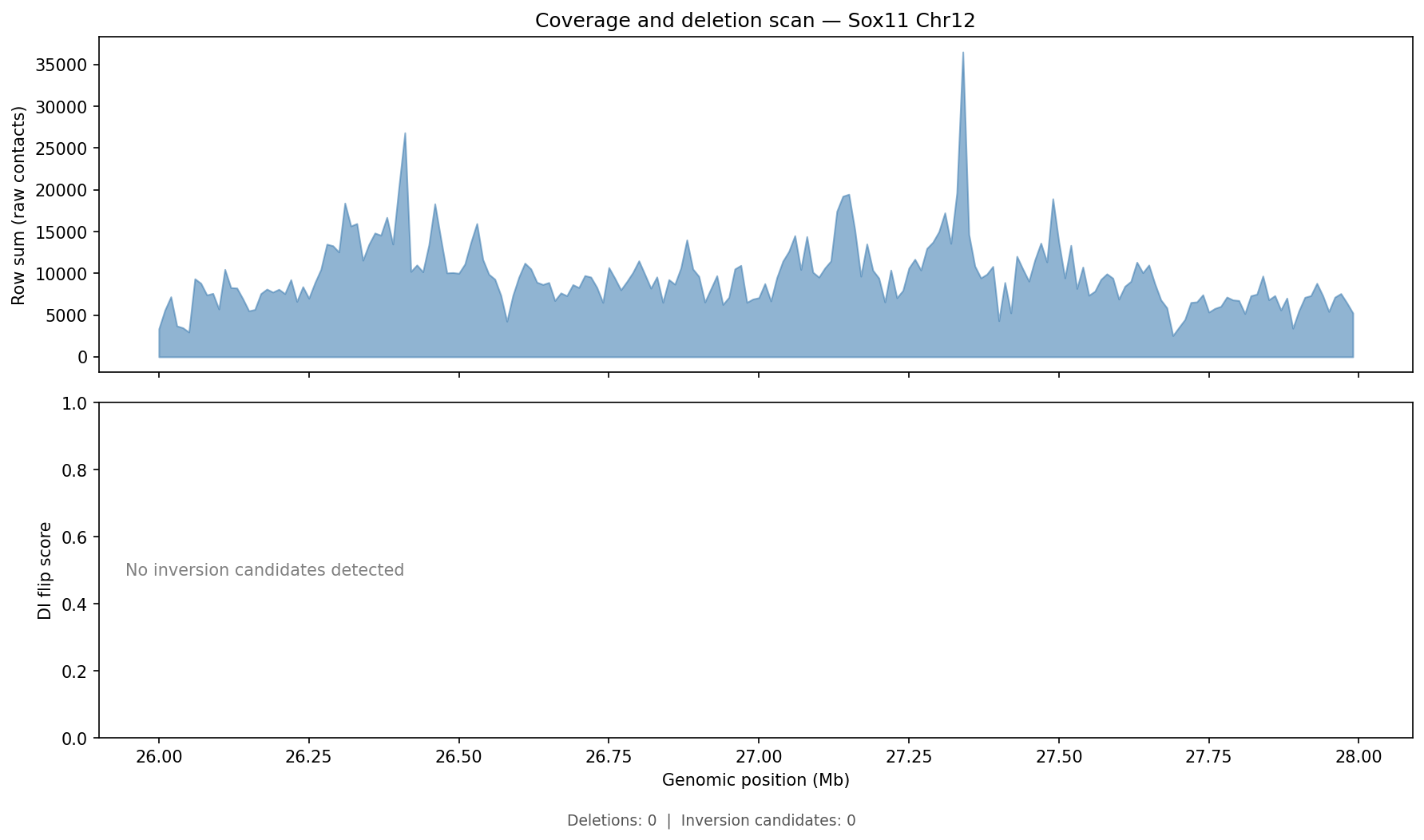
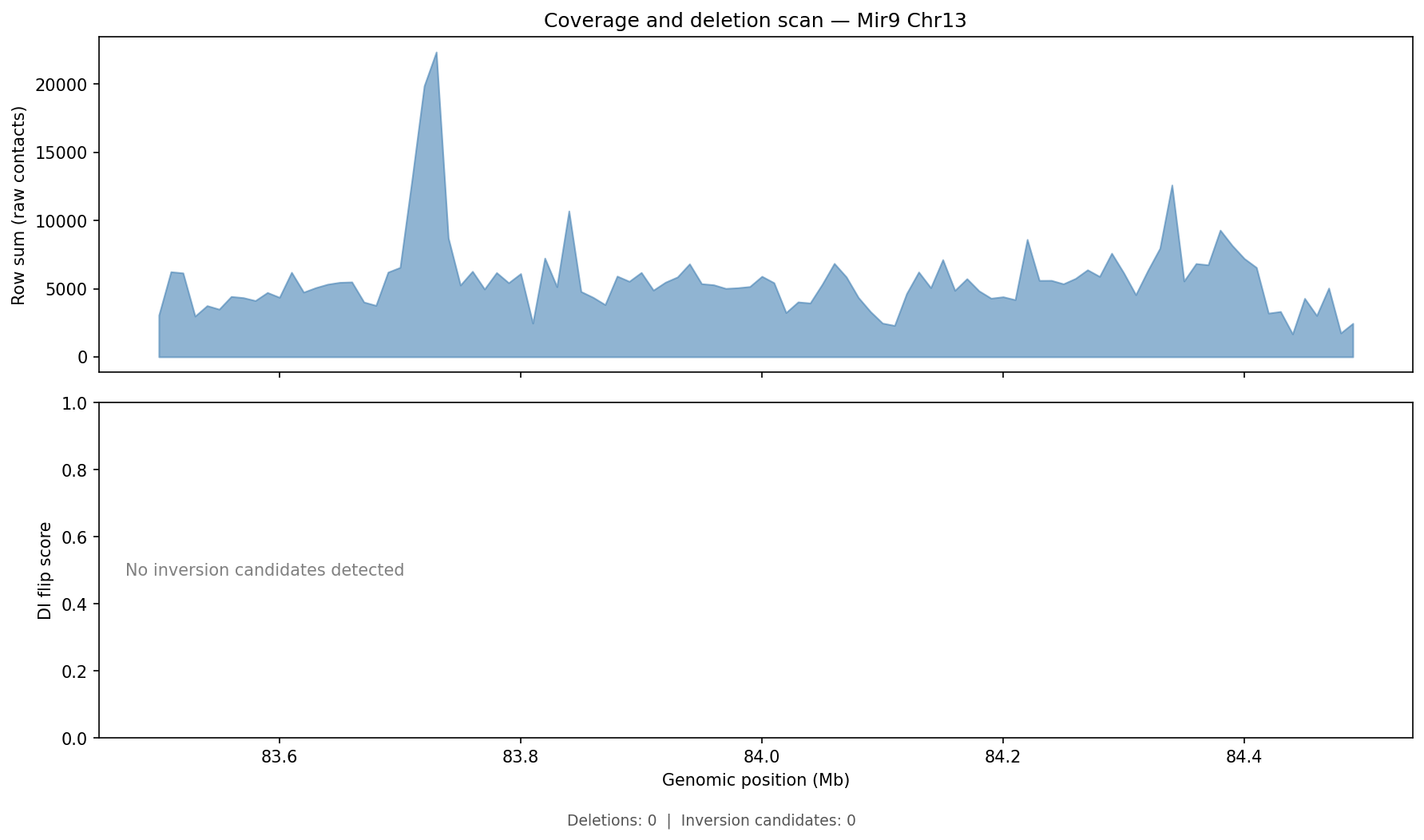
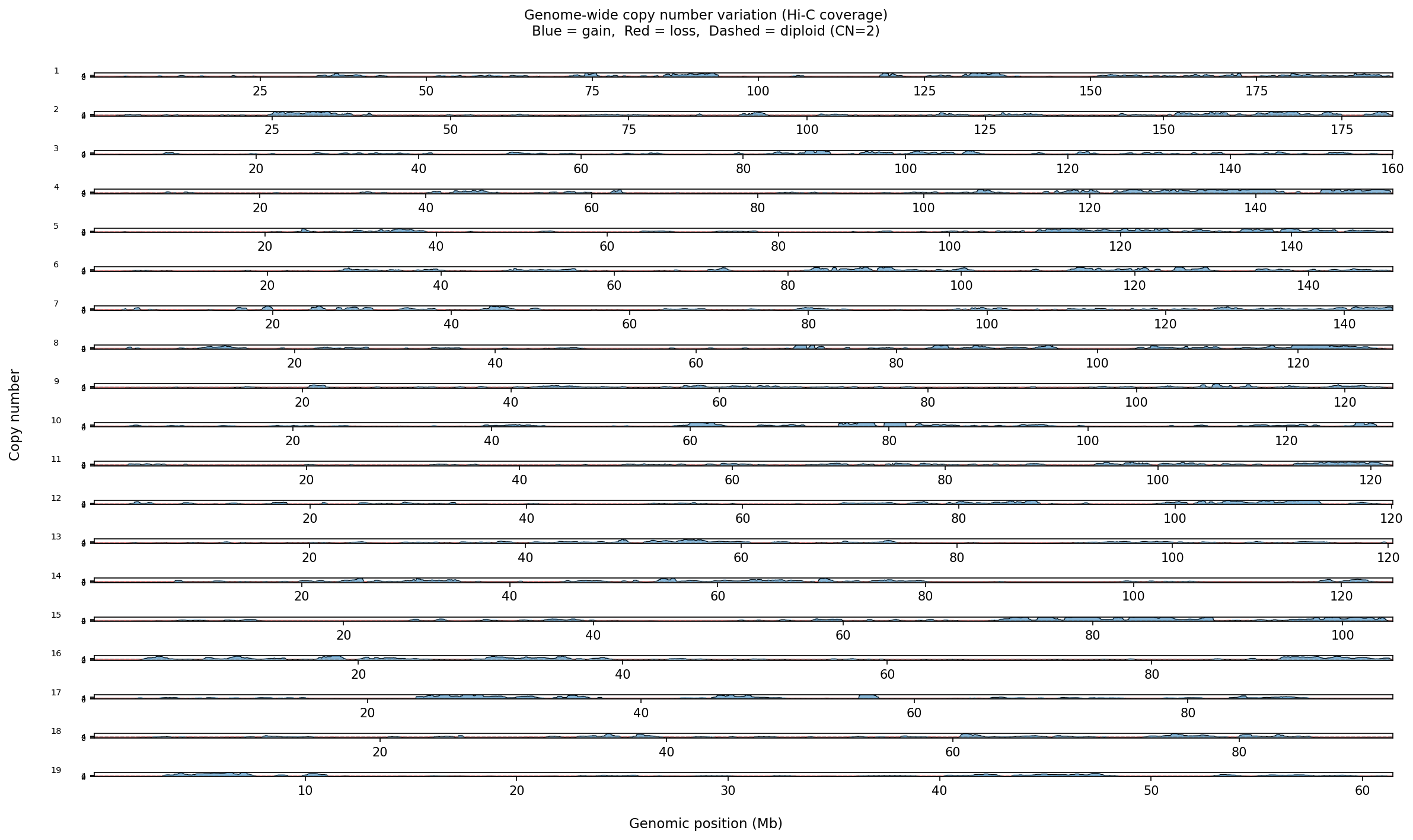
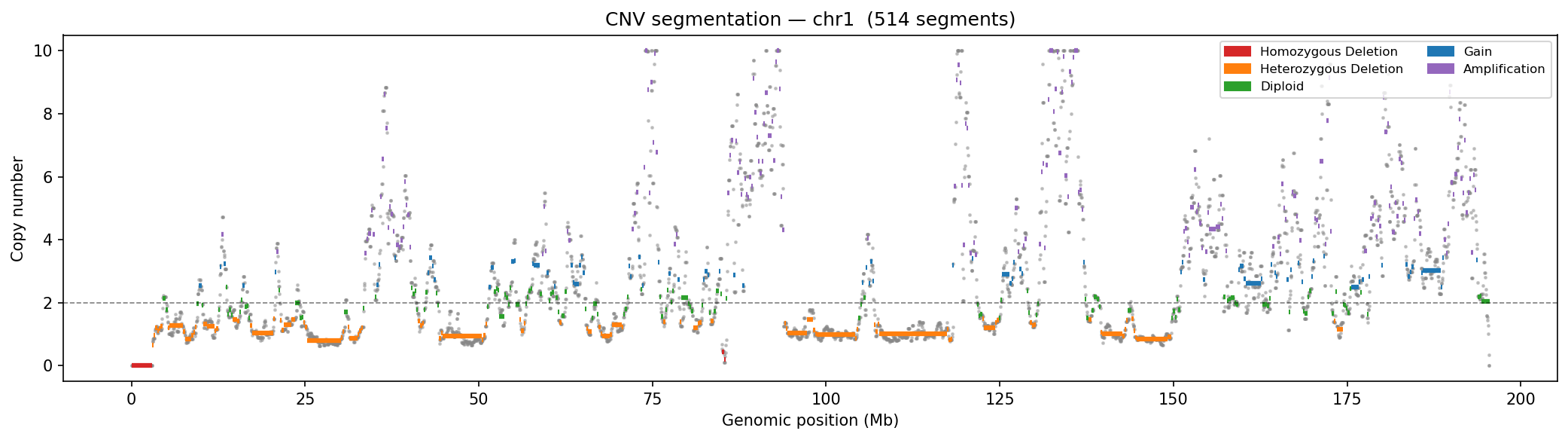
Haplotype Phasing Graph-Based
Hi-C long-range contacts link heterozygous SNPs across megabase distances, enabling chromosome-scale phasing without long reads. Heterozygous SNPs from a VCF are grouped into haplotype blocks using Hi-C read-pair connectivity: two SNPs are placed in the same block if a sufficient number of read pairs span both positions and consistently support the same haplotype assignment (graph-based phasing). Typical N50 for Hi-C phasing is chromosome-scale (Mb range) with sufficient sequencing depth. Input: heterozygous SNPs from VCF + aligned BAM.
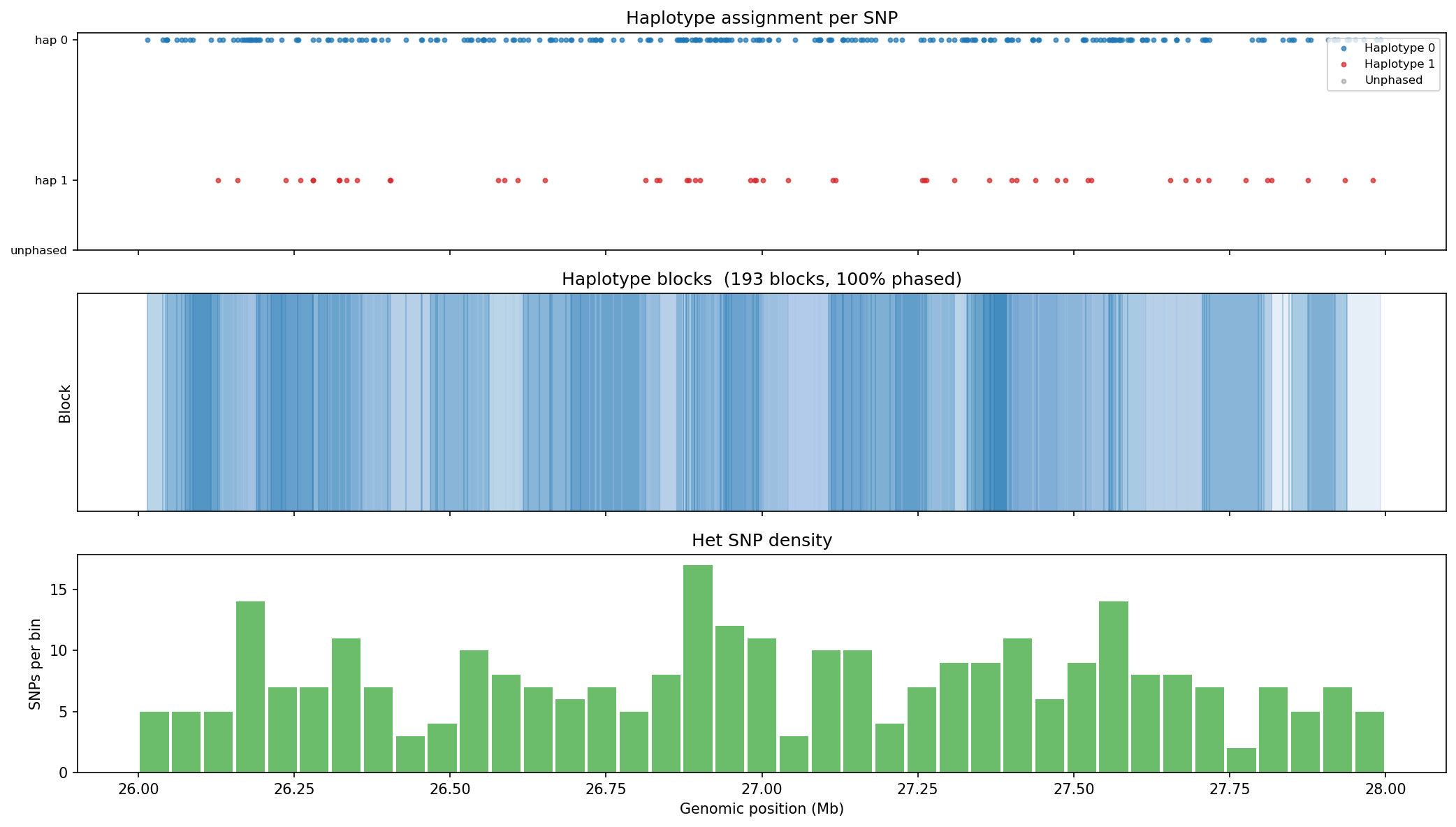
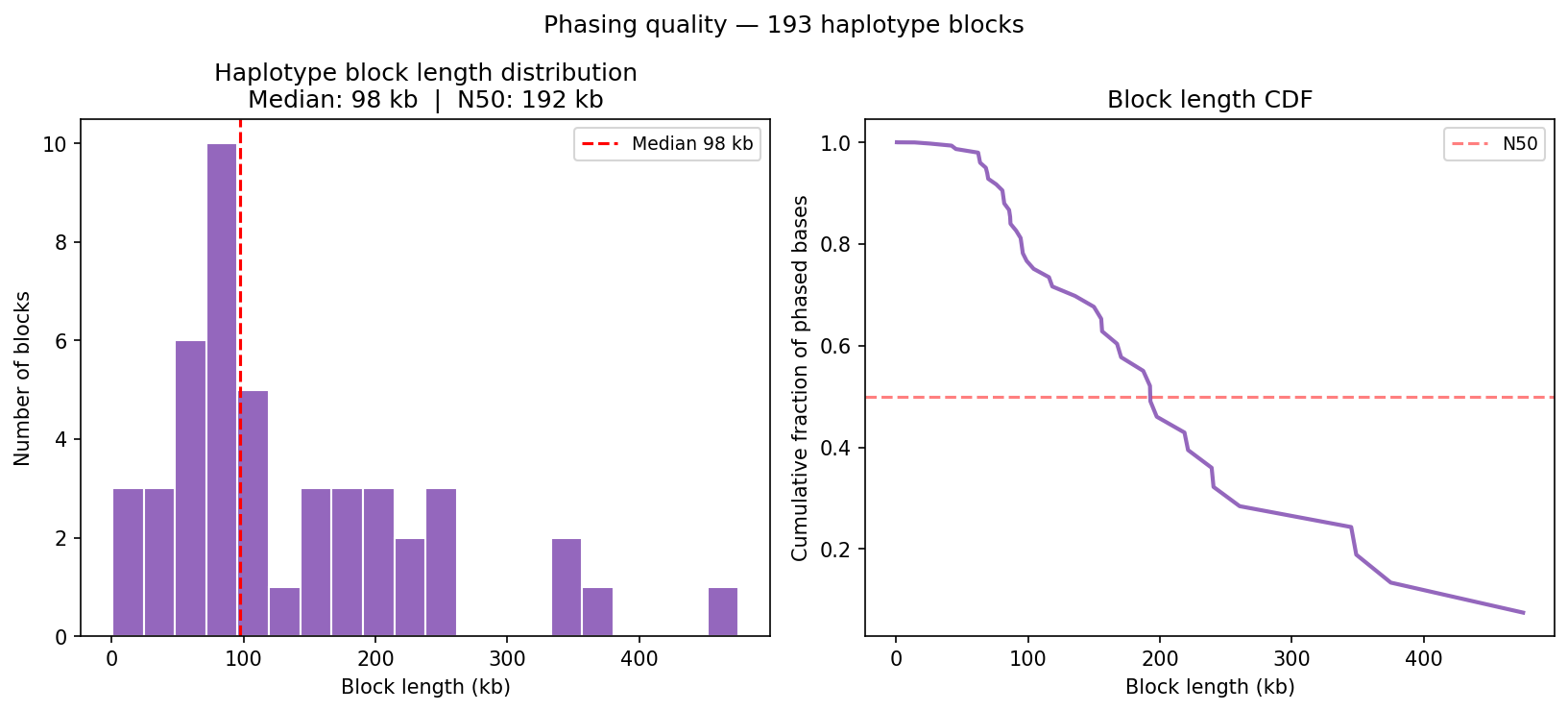
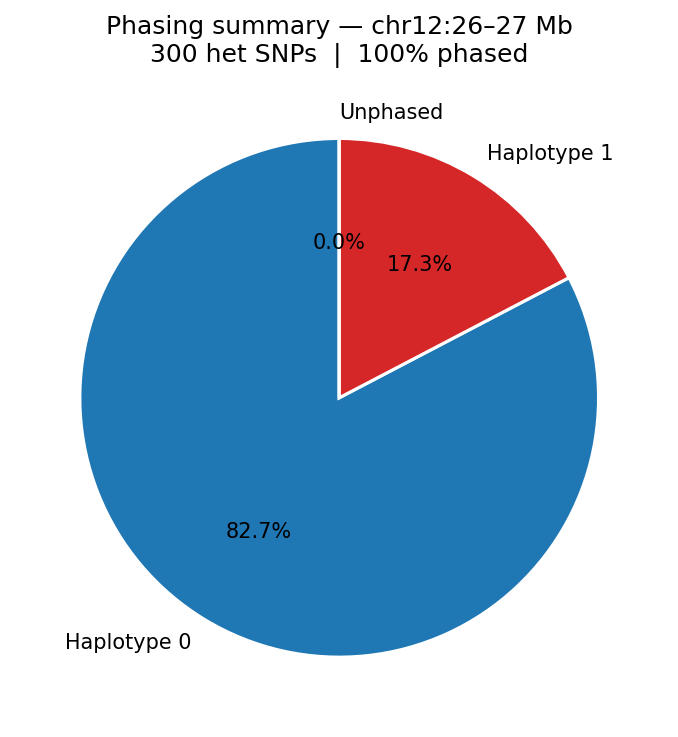
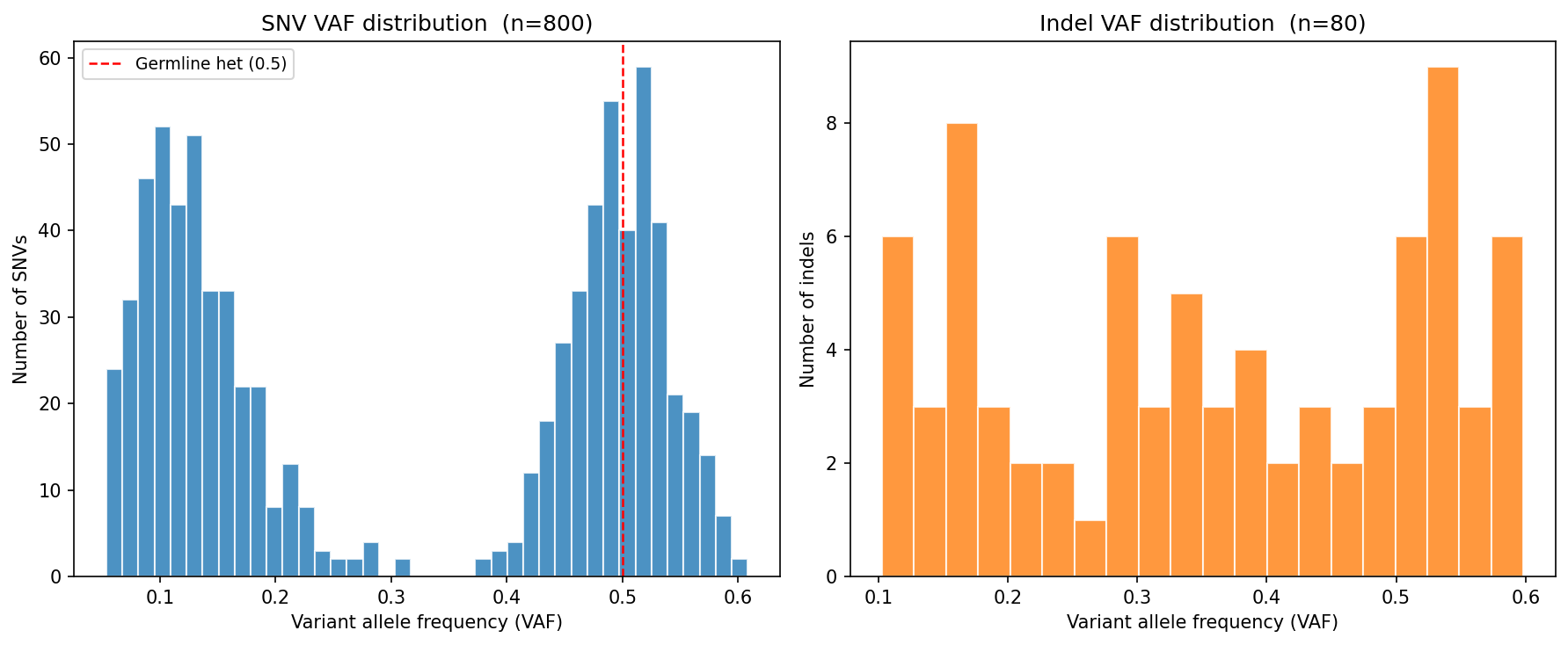
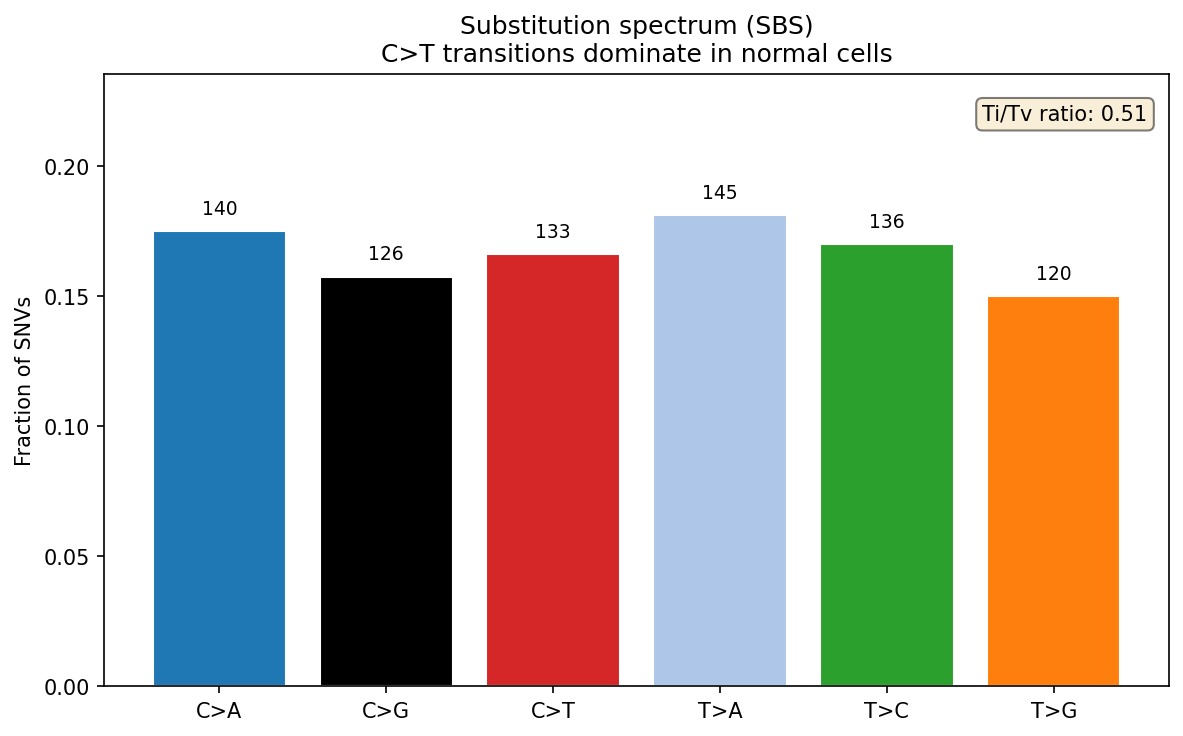
Variant Analysis SNV / Indel
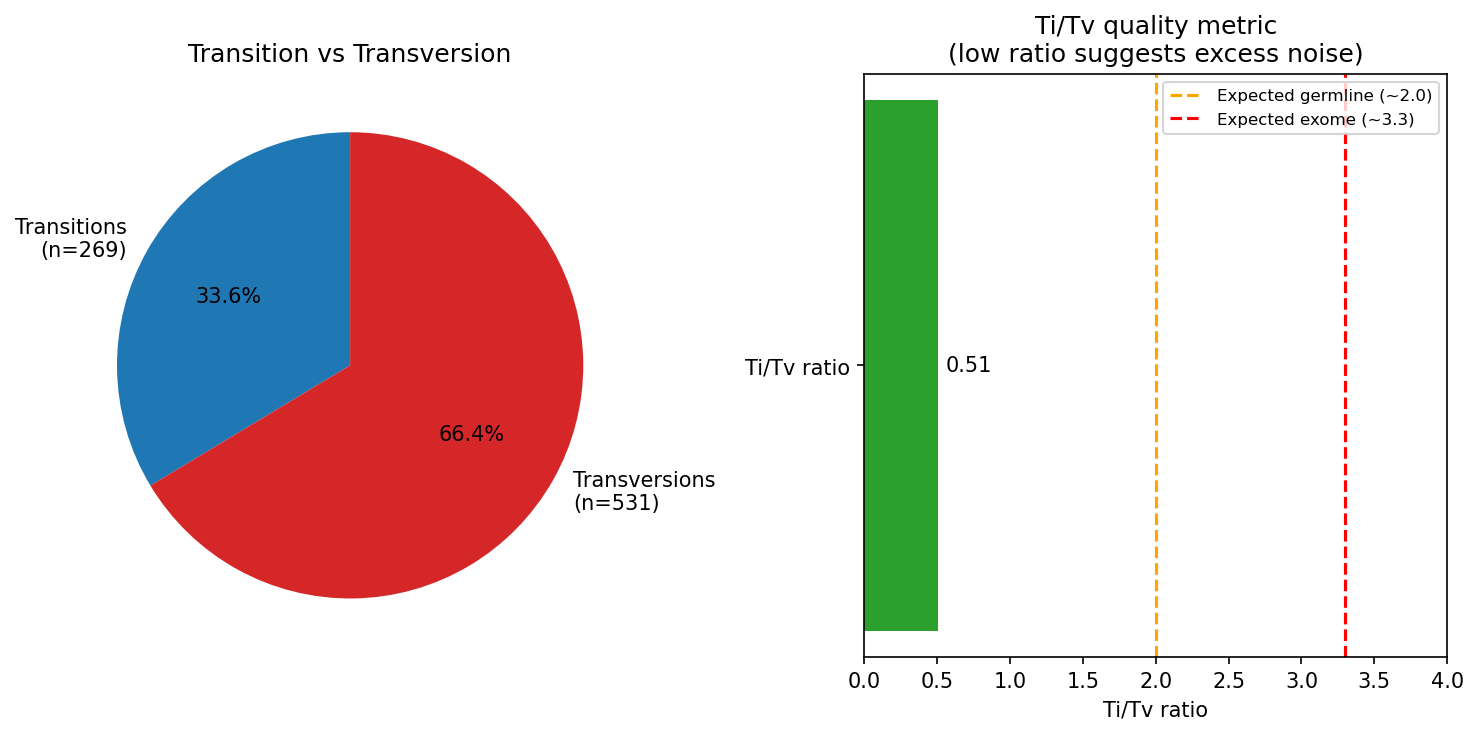
SNV and indel calling from Hi-C-aligned reads. Variant spectrum analysis characterizes mutational signatures: C>T transitions (spontaneous deamination) dominate in normal genomes; APOBEC-driven patterns (C>T/G in TC context) are elevated in certain cancers; UV signature shows C>T at dipyrimidines. Ti/Tv ratio (transition to transversion) is expected ~2.0 genome-wide and ~3.3 in exomes; values below 1.5 suggest sequencing noise or overly relaxed variant filters. Variant allele frequency (VAF) distribution distinguishes germline heterozygous variants (VAF ~0.5) from somatic mosaic variants (VAF 0.05–0.3).
How to Run on Real Data
Activate the hic-analysis conda environment and follow the steps below.
The preprocessing script follows the Dovetail recommended parameters exactly.
An AlphaGenome API key is required only for visualize_alphagenome.py.
# Prerequisites (Dovetail recommended tools)
conda activate hic-analysis
# Step 1: Preprocess FASTQ → .mcool
# Following Dovetail: BWA-MEM -5SP -T0 | pairtools parse --min-mapq 40
bash scripts/preprocess_microc.sh reference.fa R1.fq.gz R2.fq.gz sample 16
# Step 2: Run QC (Dovetail get_qc.py compatible metrics)
# Threshold: cis ≥1kb > 40% of no-dup reads
python -c "import cooler; clr = cooler.Cooler('sample.mcool::resolutions/5000'); print(clr.info)"
# Step 3: Run all analyses
python visualize_compartments.py # 25 kb, E1 eigenvector
python visualize_loops.py # 5 kb, donut background model
python analyze_hichip.py # HiChIP, requires ChIP-seq peaks BED
python analyze_capture_hic.py # Capture Hi-C, requires baits BED
python analyze_sv_cnv.py # 50 kb, genome-wide
python analyze_phasing.py # Requires het SNP VCF
python analyze_variants.py # Requires variant VCF