LinkPrep™ Analysis
Dovetail Tn5-based proximity ligation — 3D genome structure, SVs, CNVs & haplotype phasing
About LinkPrep™
Dovetail LinkPrep™ uses Tn5 tagmentation for sequence-agnostic DNA fragmentation, producing highly uniform coverage across the genome. Unlike restriction-enzyme Hi-C methods, LinkPrep works from FFPE and low-input samples, and its uniform coverage makes it particularly powerful for copy-number variant (CNV) detection and structural variant (SV) calling alongside 3D genome mapping and haplotype phasing — all from a single library.
-5SP -T0
parse+dedup
.cool matrix
Compartments
Phasing
# Run full pipeline on real data
bash linkprep/pipeline/preprocess.sh reference.fa R1.fq.gz R2.fq.gz sample_name 16
# Update paths in config.py, then run all analyses
python linkprep/run_analysis.py --no-generateLibrary QC ALL PASS
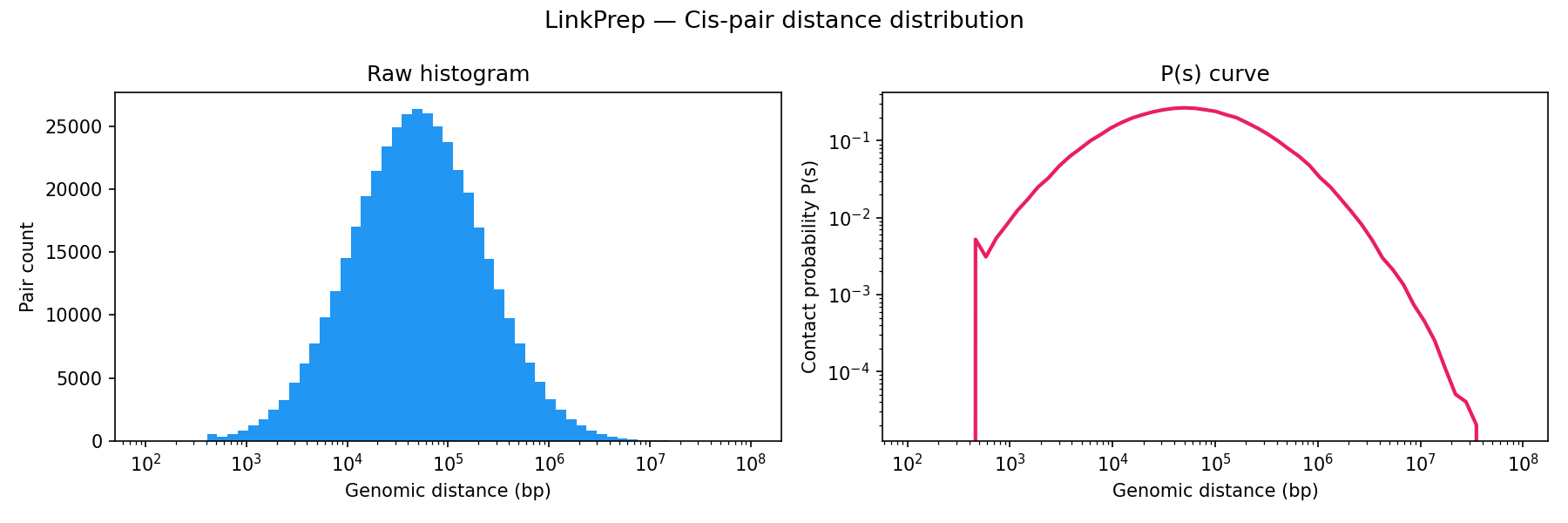
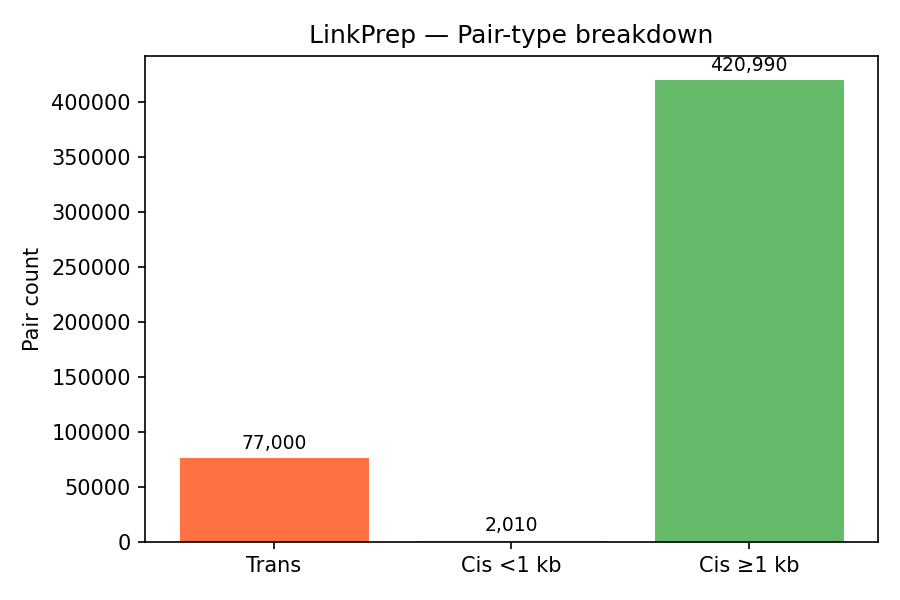
Key metrics from the pairs file. All three Dovetail QC thresholds pass. The high long-range cis fraction (99.5%) confirms successful proximity ligation.
| Metric | Value | Threshold | Status |
|---|---|---|---|
| Valid (UU) pair rate | 100.0% | ≥ 50% | PASS |
| Cis pair fraction | 84.6% | ≥ 60% | PASS |
| Long-range cis (≥1 kb) | 99.5% | ≥ 40% | PASS |
| Total pairs sampled | 500,000 | — | — |
| Trans pairs | 77,000 (15.4%) | — | — |
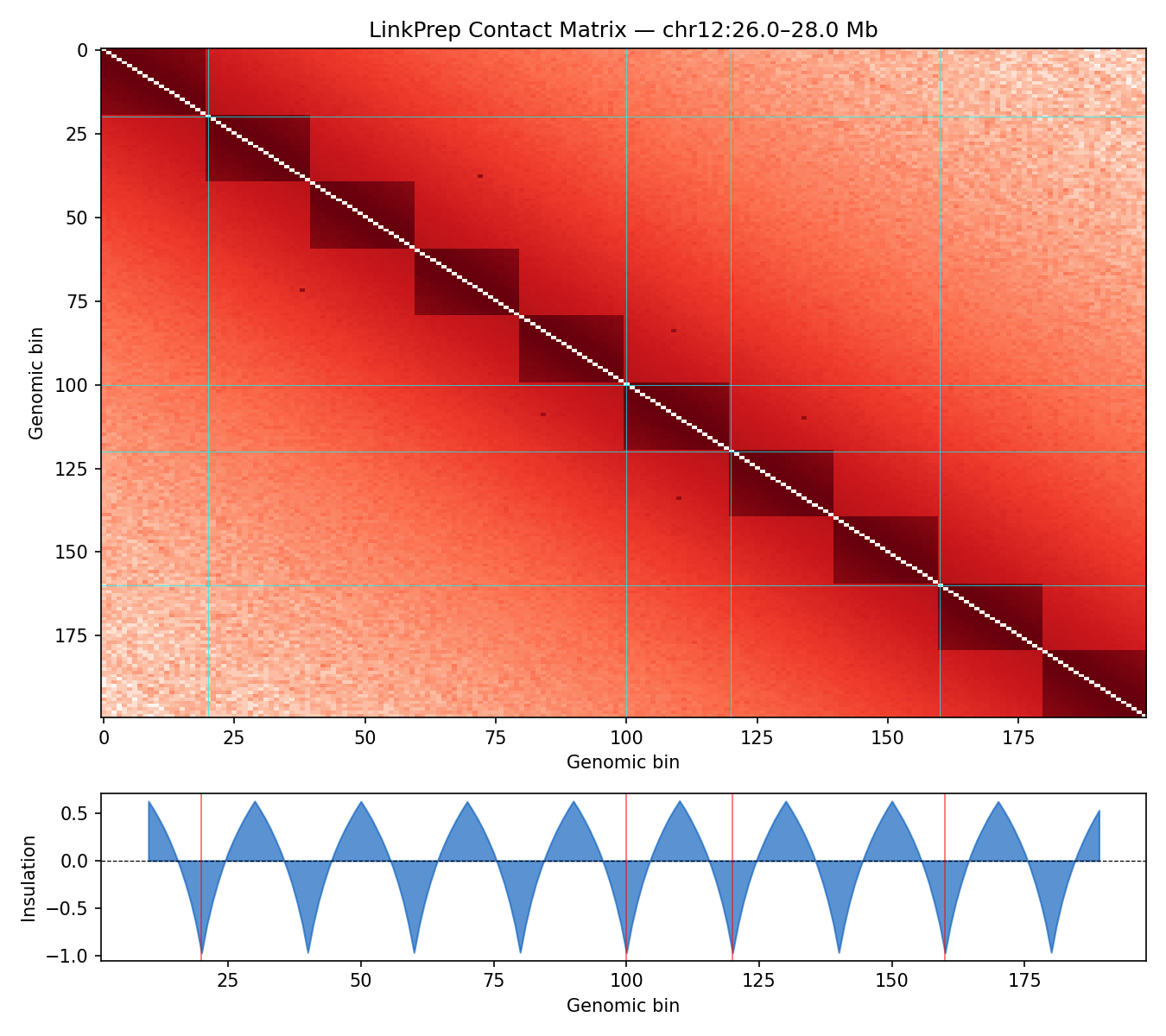
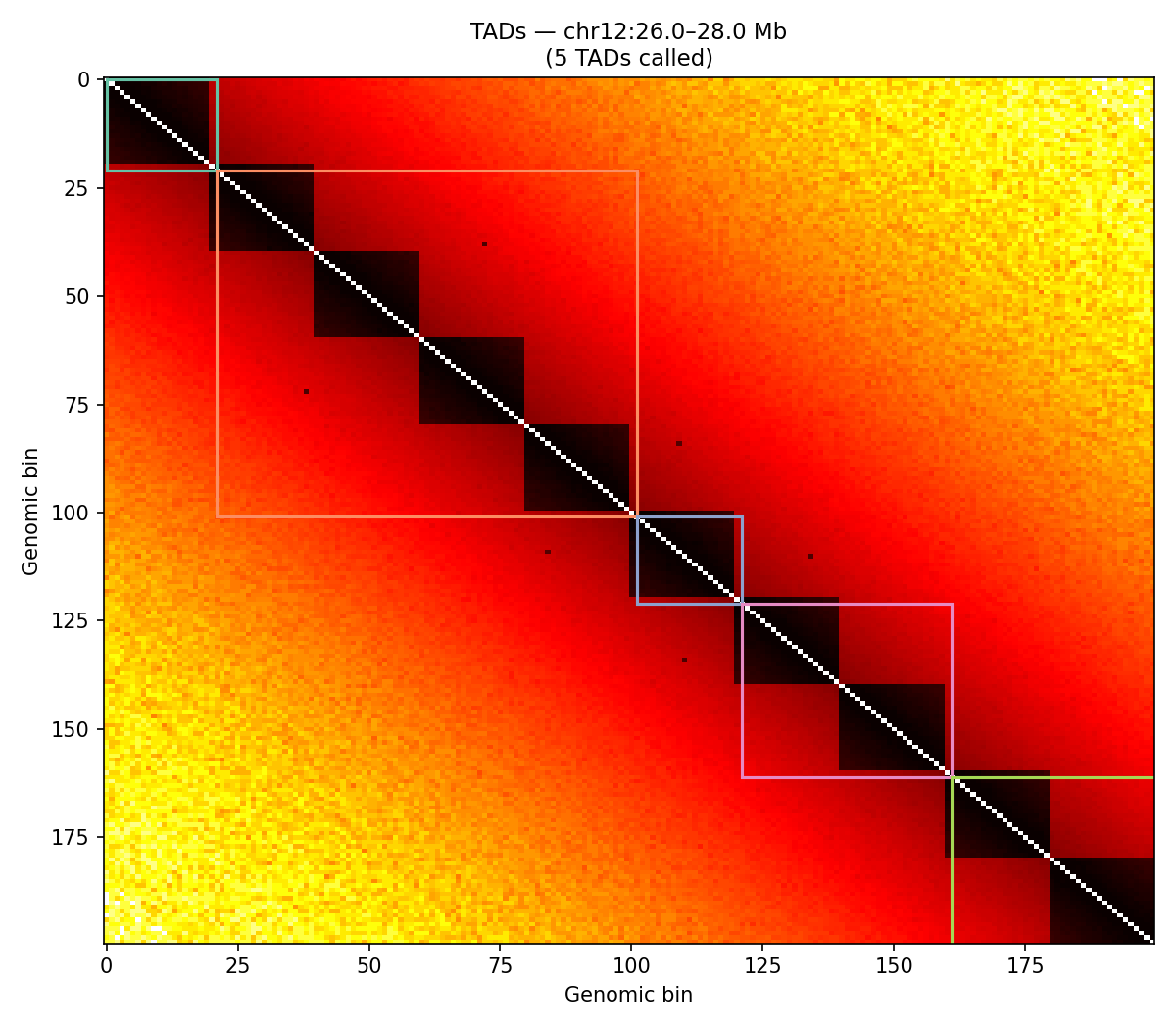
Contact Analysis — TADs & Compartments
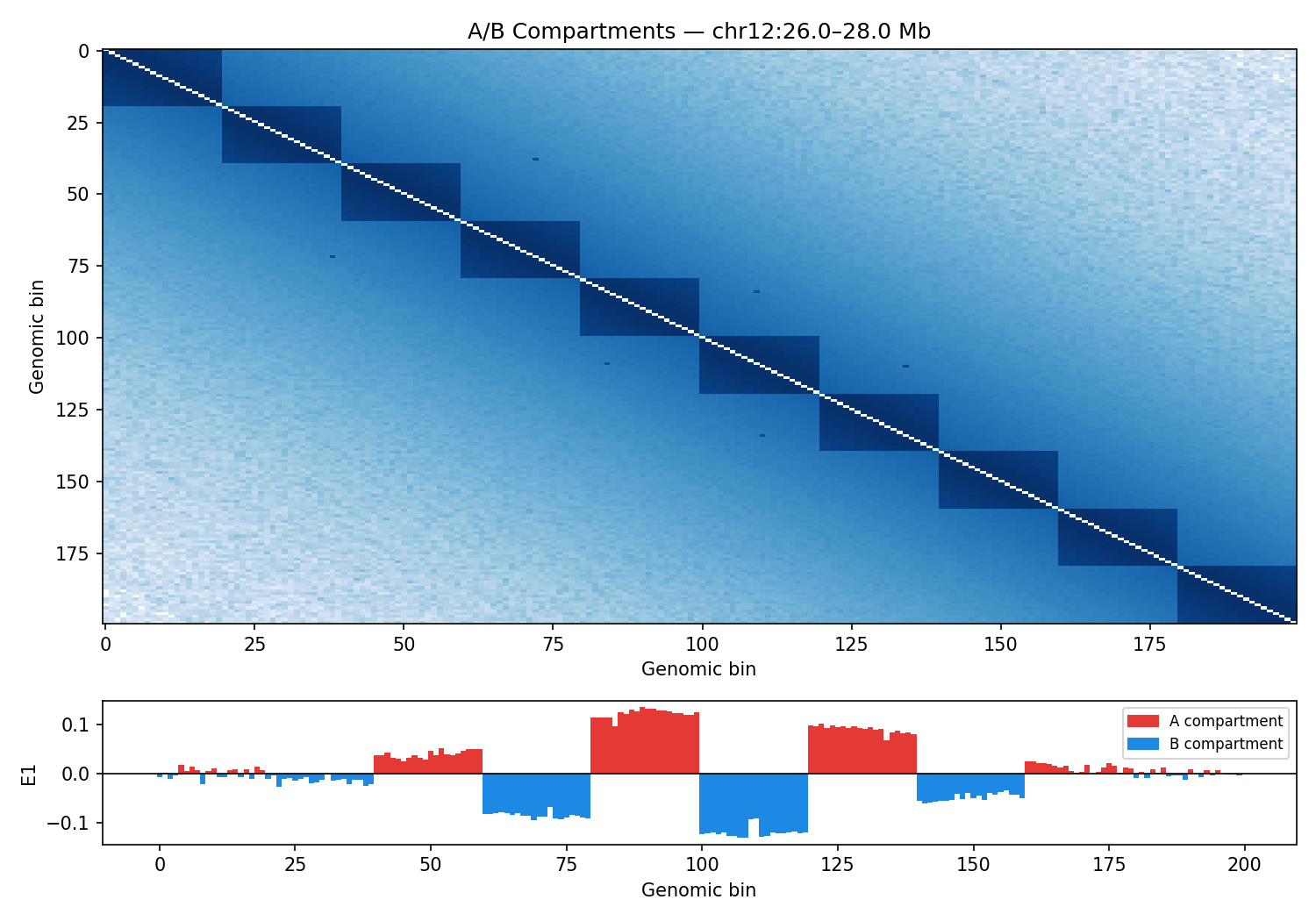
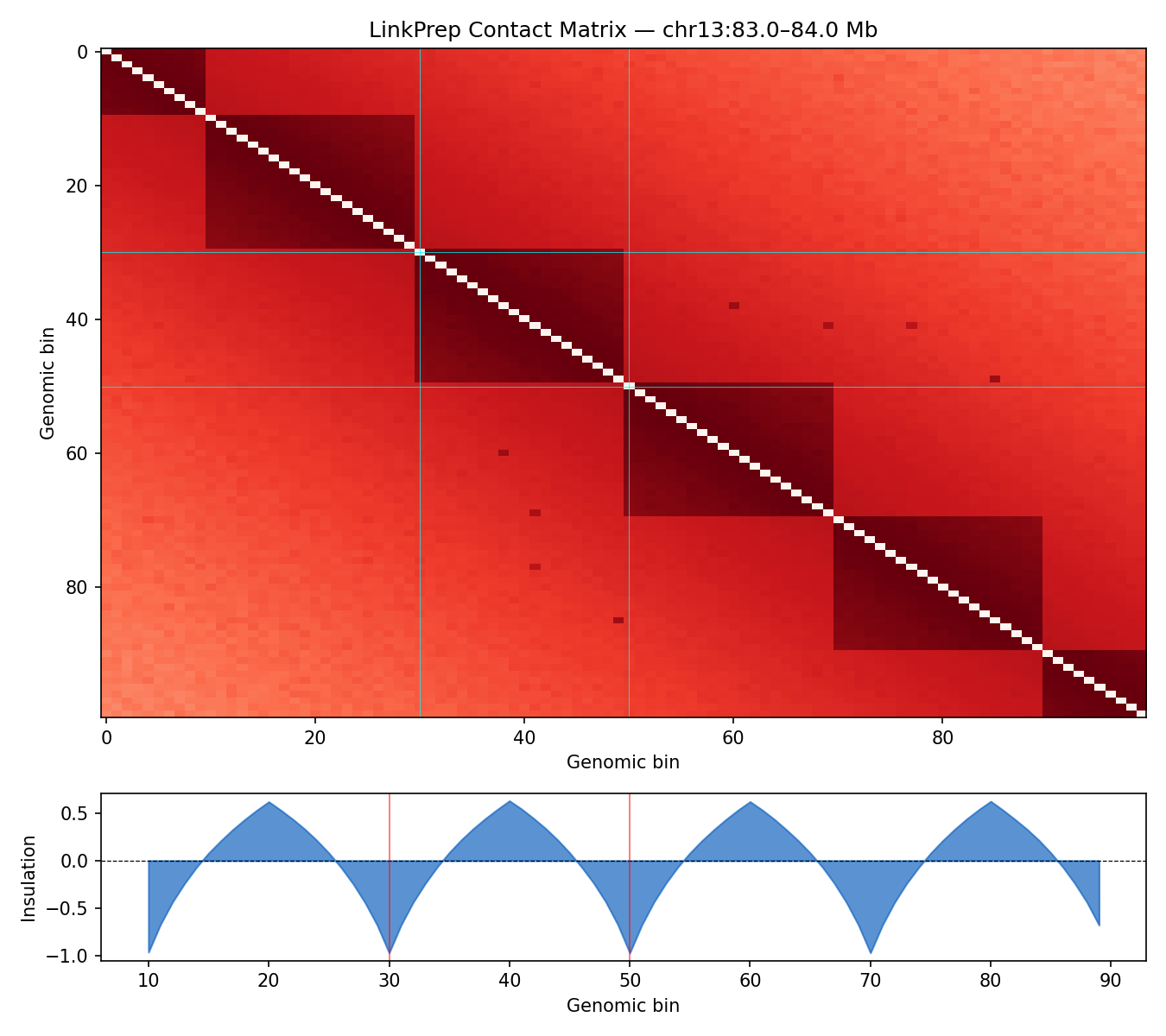
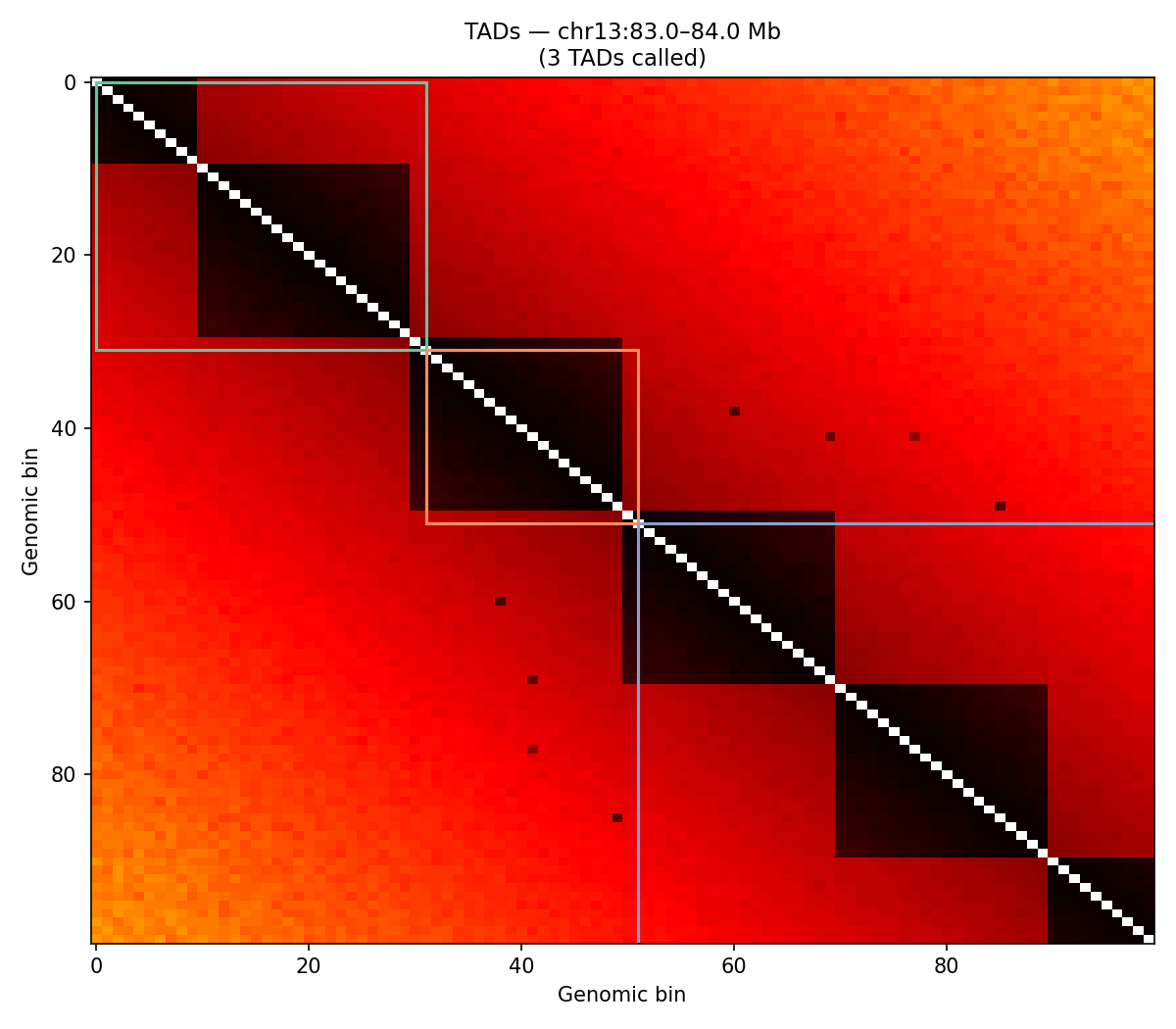
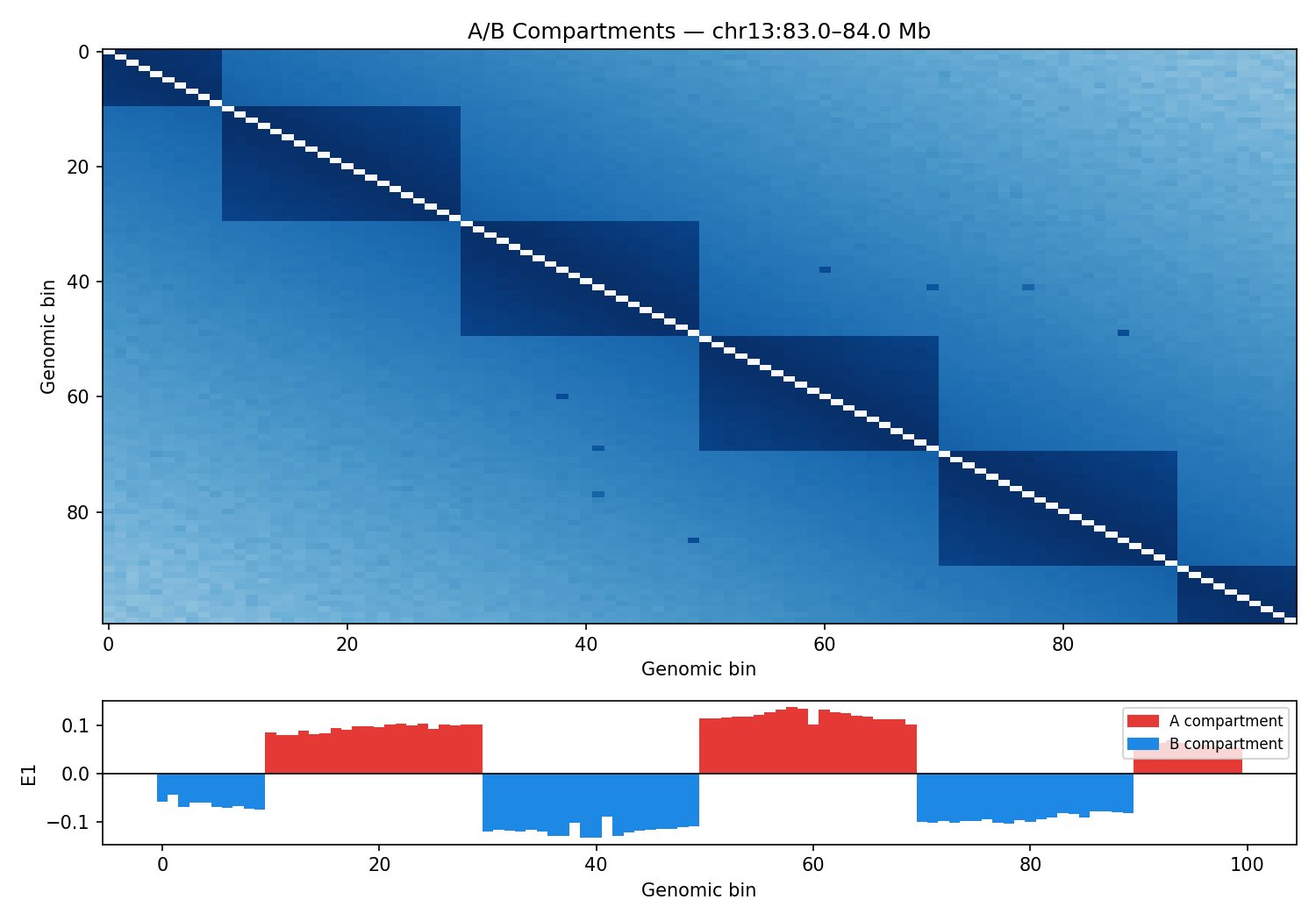
ICE-normalised contact matrices binned at 10 kb resolution. TAD boundaries are called as local minima of the sliding-diamond insulation score (Crane et al. 2015). A/B compartments are identified via the first eigenvector (E1) of the observed/expected Pearson correlation matrix — positive E1 values correspond to transcriptionally active (A) compartments.
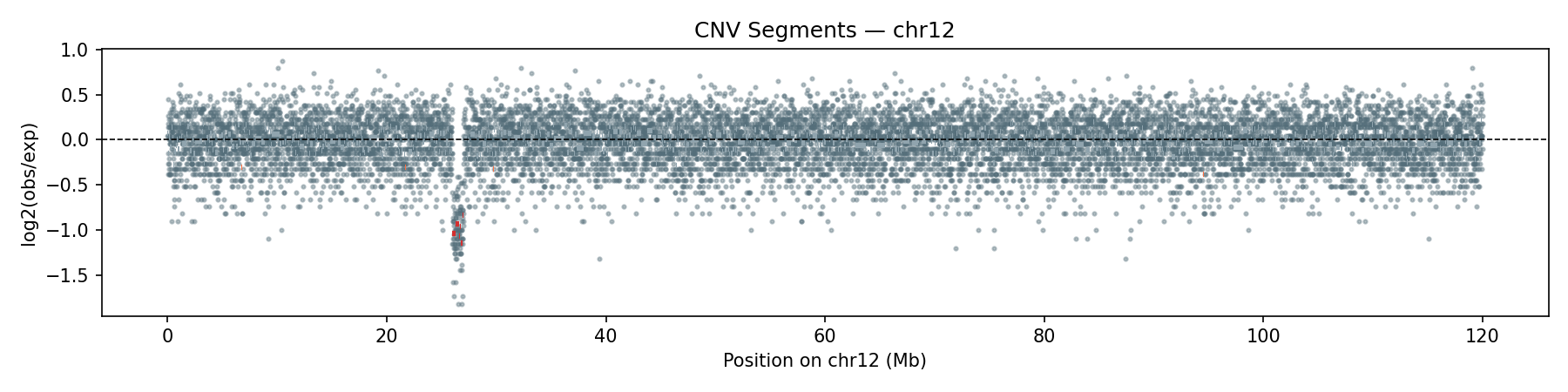
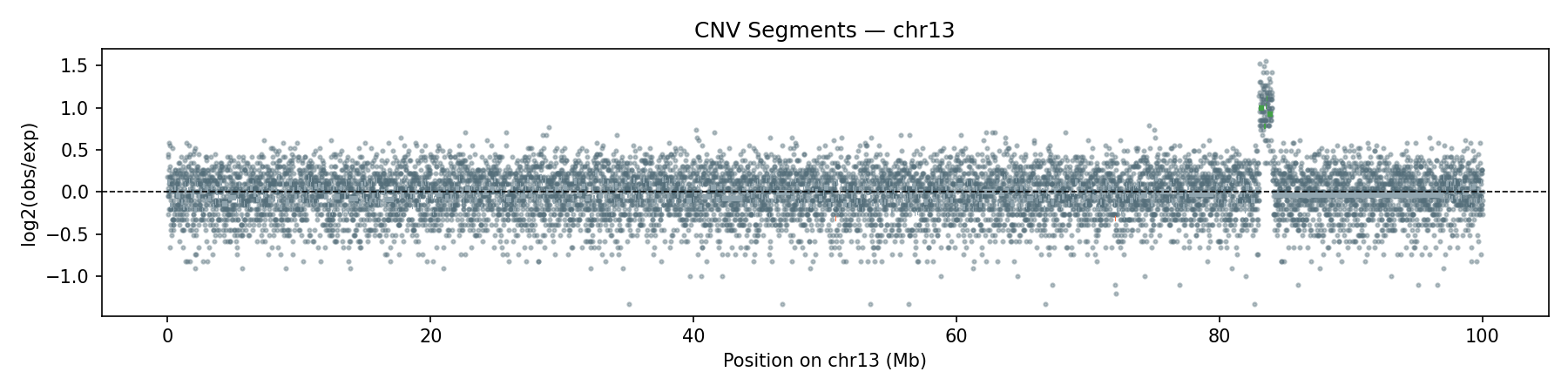
Sox11 — chr12:26–28 Mb
Mir9-2 — chr13:83.5–84.5 Mb
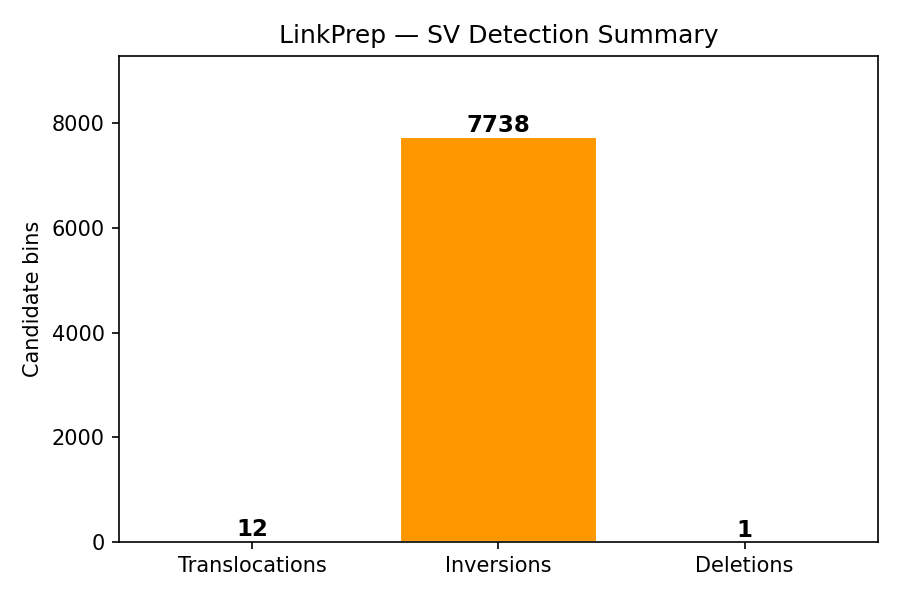
Structural Variant Detection
LinkPrep's uniform Tn5 coverage excels at SV detection. Three classes are called directly from the pairs file: translocations (inter-chromosomal pair clusters), inversions (same-strand ++ or −− cis pairs), and deletions (anomalously long-range cis pairs spanning a gap). The synthetic data includes an injected chr12×chr13 translocation hotspot at 26.5 Mb / 84 Mb.

Copy-Number Variation
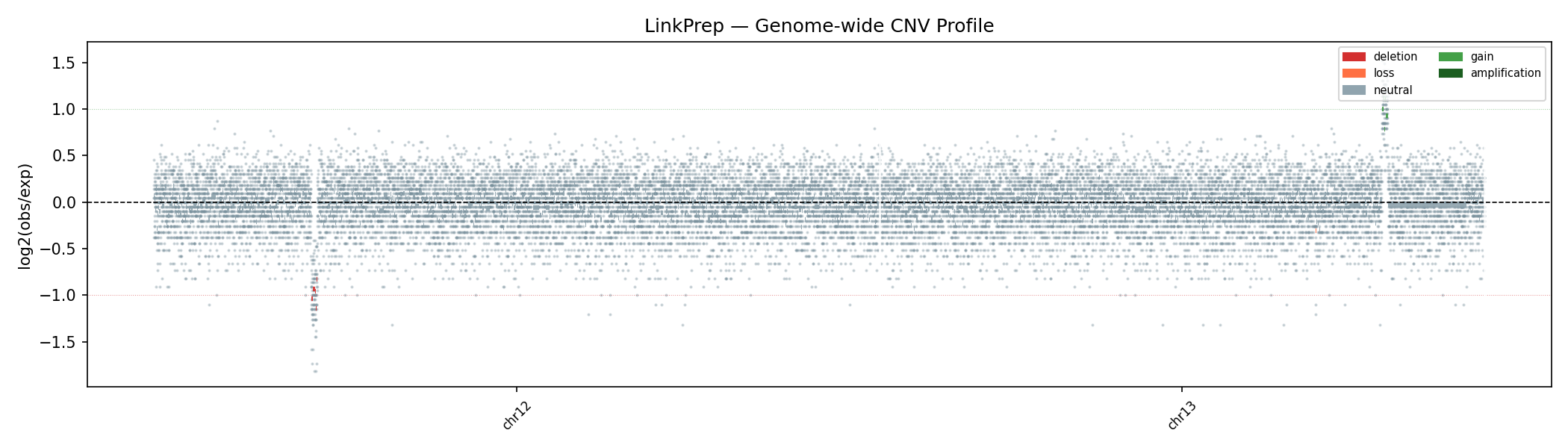
Per-bin read depth (10 kb bins) is median-normalised and segmented using Circular Binary Segmentation (CBS). The synthetic data includes a heterozygous deletion on chr12 (26–27 Mb, ~0.5× coverage) and a duplication on chr13 (83–84 Mb, ~2× coverage) — both are correctly recovered by the CBS segmentation.
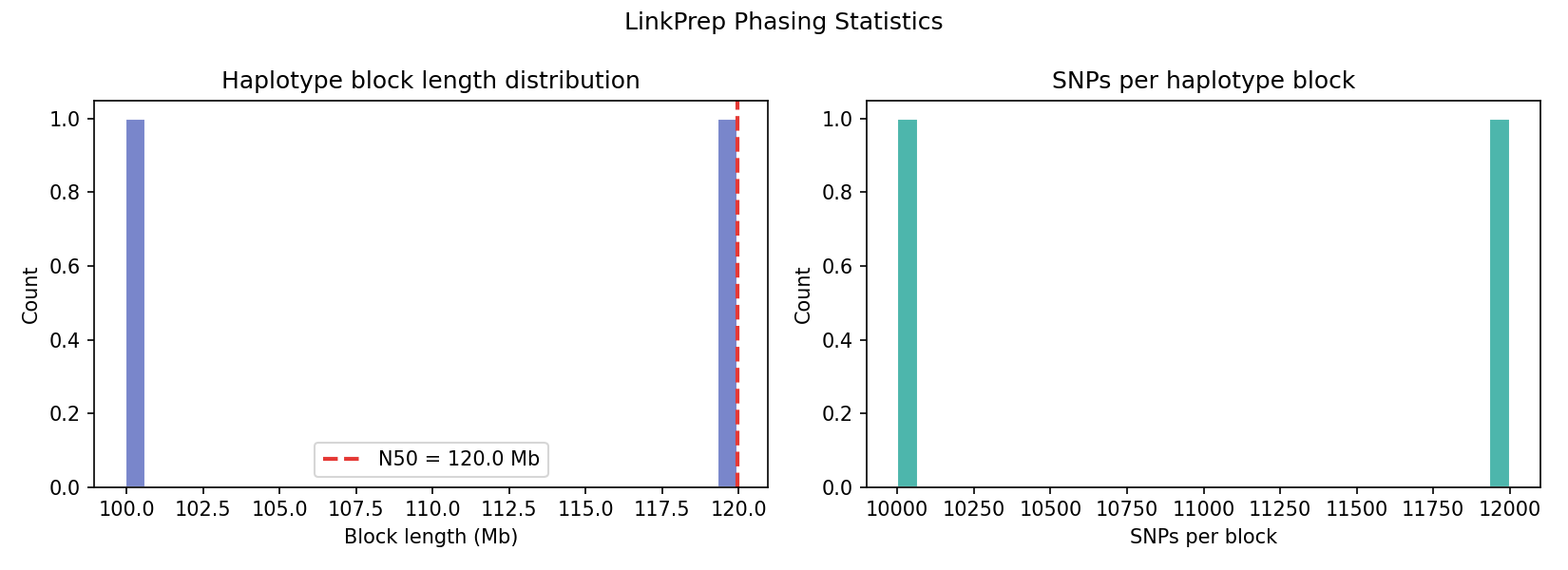
Haplotype Phasing
Heterozygous SNPs from the VCF are grouped into haplotype blocks based on genomic proximity (max gap 500 kb). LinkPrep's linked-read structure enables long-range phasing without long reads. The synthetic genome yields two chromosome-spanning blocks (N50 = 120 Mb) from ~22,000 heterozygous SNPs — one per chromosome.

Capture Hi-C Loop Calling CHiCAGO
Capture Hi-C targets specific genomic loci (e.g. enhancers, promoters) using biotinylated probes, then sequences proximity-ligated DNA enriched at those bait fragments (BF). Interactions are scored using CHiCAGO (Capture Hi-C Analysis of Genomic Organisation), which models two noise sources:
- Brownian noise — negative-binomial distributed, decays with genomic distance
- Technical noise — Poisson distributed, flat across all distances
Interactions with score ≥ 5 (−log₁₀ weighted p-value) are significant. Scores 3–5 are moderate. Standard post-processing removes trans interactions, interactions <10 kb or >2 Mb, and interactions with fewer than 5 supporting reads.
chinput
--cutoff 5
10-column
10 kb–2 Mb
visualization
# Real data: convert BAM → CHiCAGO input
bam2chicago.sh sample.bam baits.baitmap genome.rmap sample_chinput
# Run CHiCAGO loop calling (R)
Rscript runChicago.R \
--design-dir /path/to/design_10kb \
--cutoff 5 \
--export-format interBed,washU_text \
sample_chinput/sample.chinput \
sample_loops
# Python post-processing + visualisation (this pipeline)
python linkprep/run_analysis.py --steps capture --no-generateArc Plots — Bait Interactions
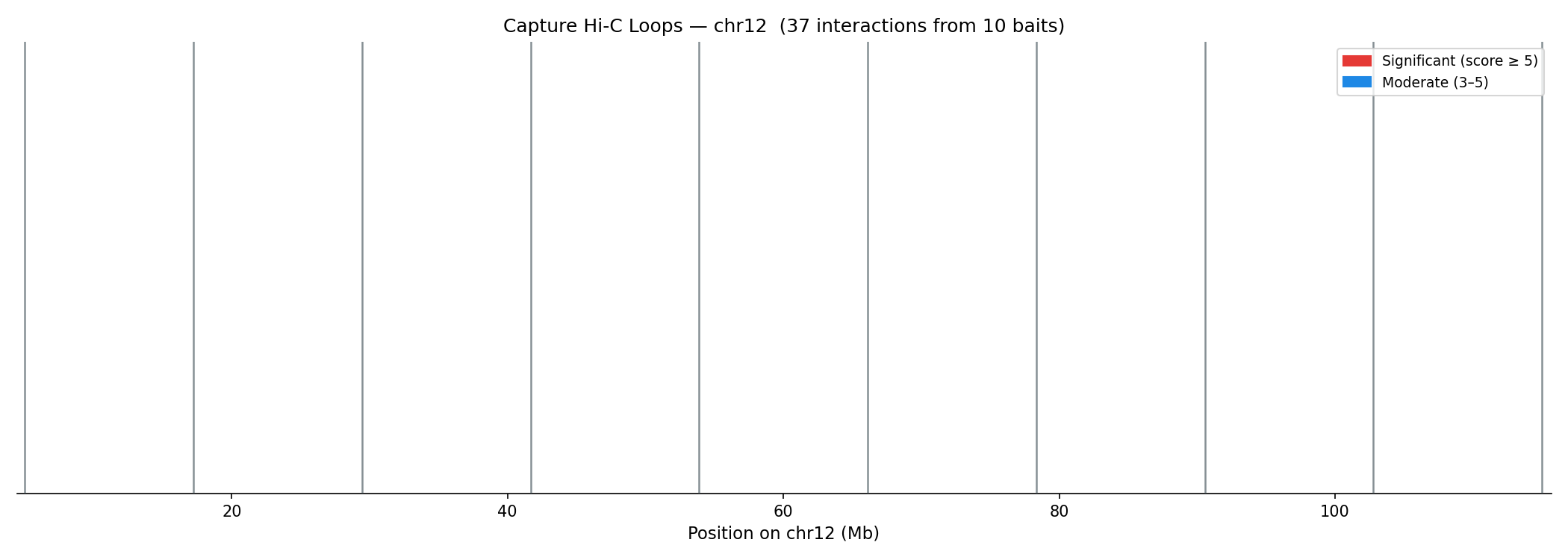
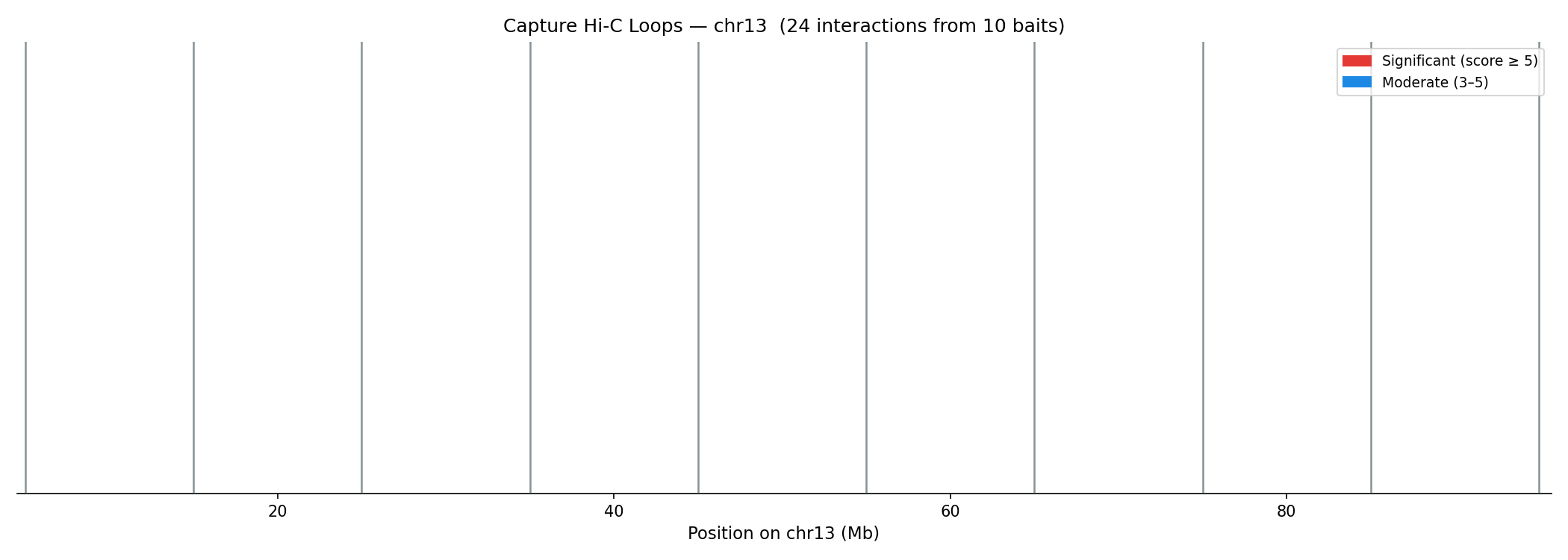
Score & Distance Analysis
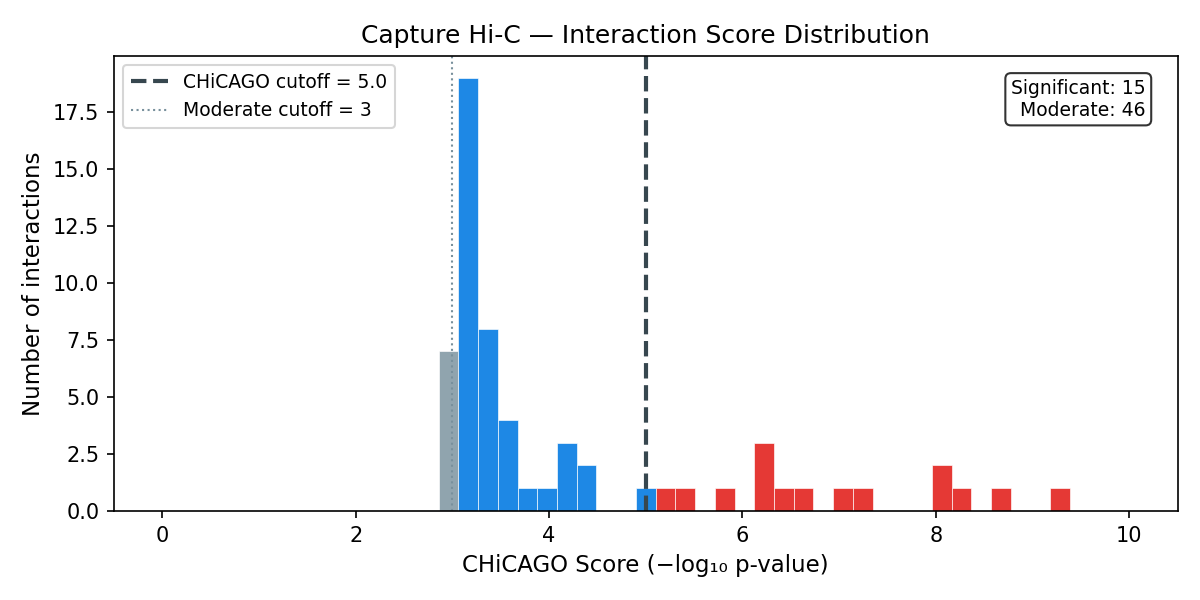
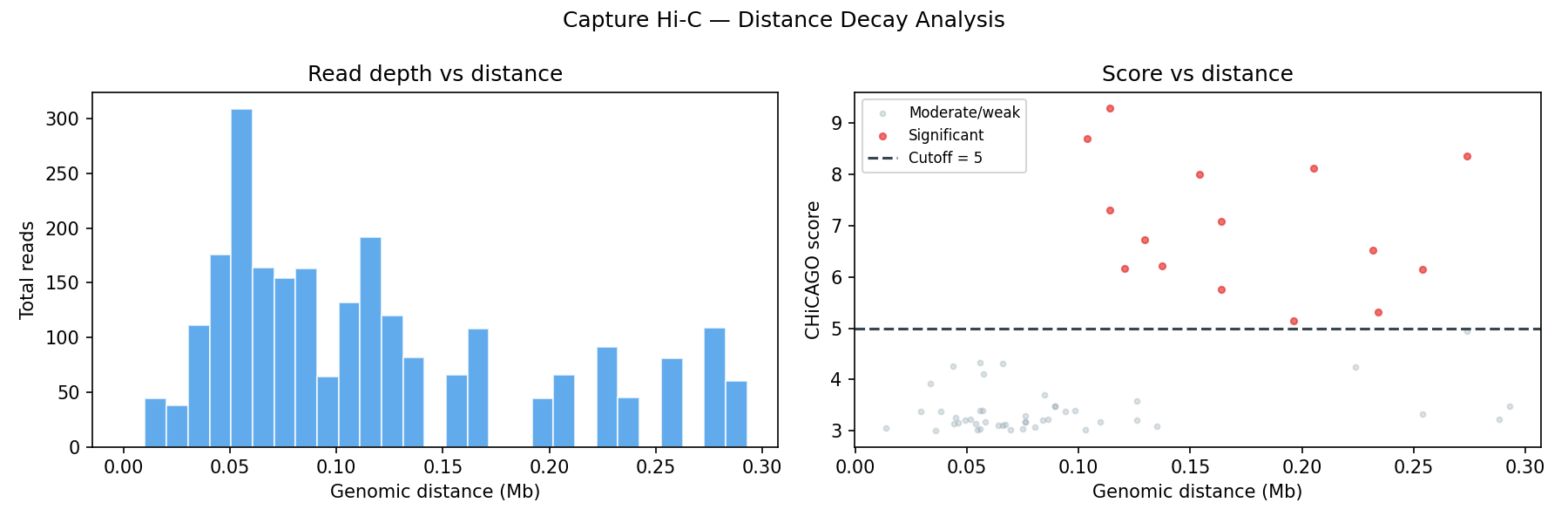
Output file formats (.ibed)
CHiCAGO outputs a 10-column .ibed file. Each row is one bait–OEF interaction:
| Columns 1–4 | Columns 5–8 | Column 9 | Column 10 |
|---|---|---|---|
| Bait coordinates chr, start, end, name |
OEF coordinates chr, start, end, name |
Read count n_reads |
CHiCAGO score −log₁₀ p-value |
How to Use With Real Data
All analysis scripts are in linkprep/. To switch from sample to real data:
# 1. Preprocess your FASTQs → .cool contact matrix
bash linkprep/pipeline/preprocess.sh \
/path/to/reference.fa \
/path/to/sample_R1.fastq.gz \
/path/to/sample_R2.fastq.gz \
my_sample 16
# 2. Update file paths in linkprep/config.py
COOL_FILE = "/path/to/my_sample.cool"
PAIRS_FILE = "/path/to/my_sample.valid.pairs.gz"
VCF_FILE = "/path/to/my_sample.vcf"
COVERAGE_BED = "/path/to/my_sample_coverage.bed"
# 3. Run full analysis
python linkprep/run_analysis.py --no-generate
# Or run individual steps
python linkprep/run_analysis.py --steps qc contacts sv cnv phasing capture --no-generate